ABSTRACT
Cystic Fibrosis (CF) is a multisystem disorder primarily impacting the lungs, characterized by bronchial obstruction, infection and inflammation. Over 75% of CF patients are diagnosed by age 2, with most living beyond 18. The disease is linked to over 2,000 mutations in the CFTR gene. Current treatments focus on CFTR modulators like ivacaftor, lumacaftor and tezacaftor, which enhance CFTR protein function and improve lung health. However, these therapies face challenges, including limited efficacy against a wide range of CFTR mutations, high costs and concerns about long-term safety and effectiveness. Poor patient adherence and inefficacy of genetic therapies further complicate treatment. This review explores the genotypic and phenotypic expression of CFTR, evaluates current treatment strategies and analyzes the clinical outcomes of CFTR modulators in specific populations. It also examines the scope of ongoing clinical trials and considers future approaches, such as advanced gene editing technologies and personalized medicine, aiming to address the limitations of current treatments and improve disease management. These futuristic strategies hold promise in enhancing therapeutic efficacy, broadening mutation coverage and reducing the prevalence of CF through more targeted and durable interventions
INTRODUCTION
Cystic Fibrosis (CF) is an autosomal recessive disorder caused by mutations in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene, affecting the respiratory and gastrointestinal systems. With 40,000 adults and children in the US and 105,000 worldwide, the disease has a sharply increasing incidence and survival rate (Rafeeq and Murad, 2017). The discovery of the CFTR gene in 1989 provided hope for the scientific and medical sectors, leading to the identification of over 2000 different mutations. This has improved diagnosis and care for CF patients and their families and opened the door for the creation of treatments specific to specific mutations. CF damages multiple organs, leading to progressive lung damage and mortality. Advancements in healthcare and treatments have raised the median age of death for CF patients to the early fifties. Recent advances in pharmaceutical interventions, particularly CFTR modulators, allow for targeting the genetic root of CF mutations, potentially rescuing mutant CFTR function and improving the quality of life for CF patients (Sui et al., 2022). This review is organized into the following sections. CFTR Genetic lesions: Discusses the spectrum of genetic mutations, including missense, nonsense and splicing defects, with a focus on their molecular mechanisms. Phenotypic Expressions of Cystic Fibrosis: Explores the correlation between specific CFTR mutations and the resulting clinical manifestations, emphasizing pulmonary, gastrointestinal and reproductive health. Current Therapeutic Approaches: Summarizes established treatment strategies, including symptomatic management and CFTR modulators. Emerging Therapies: Highlights recent advancements in gene therapy, mRNA therapy and small-molecule correctors targeting underlying genetic defects. Challenges and Future Directions: Discuss existing gaps in CF management and research and outline potential strategies for overcoming these barriers. Conclusion: Provides a concise summary of findings and their implications for clinical practice and research.
CFTR GENETIC LESIONS
CFTR Gene and Protein Structure
Cystic Fibrosis (CF) is a genetic disorder characterized by the presence of two copies of CFTR-mutated genes from both parents. The CFTR gene is located on chromosome 7 and is composed of 27 coding exons. Over 2,000 distinct CFTR gene variants have been reported, with 432 found by the Clinical and Functional Translation of CFTR. Some variants have shown disease liability and clinical consequences. The CFTR protein is a 1,480-amino acid protein located on the apical membrane of epithelial cells. It is composed of five distinct domains: Transmembrane Domains (TMD1 and TMD2), Nucleotide-Binding Domains (NBD1 and NBD2) and a Regulatory Domain (RD). The CFTR protein’s biosynthesis and post-translational folding are significantly influenced by the TMDs. The CFTR protein’s complex structure allows chloride ions to exit epithelial cells and it is expressed in various organs such as the lungs, upper respiratory tract, pancreas, liver, gallbladder, intestines and sweat (Pereira, Ribeiro, Ribeiro, Bertuzzo, and Marson, 2019; Liu et al., 2019; Zhang, Liu, and Chen, 2017).
Types of CFTR Mutations
Class I CFTR mutations result in a shortened, non-functional CFTR protein, leading to recurrent infections, inflammation and lung damage. These mutations are the most severe type, accounting for 5-10% of cases of cystic fibrosis. Notable nonsense mutations include G542X, W1282X and R553X. Class II mutations cause the CFTR protein to misfold, preventing it from reaching the cell membrane. The most common mutation, F508del, causes the phenylalanine at position 508 of the CFTR protein to be deleted, causing the protein to misfold and disrupt the three-dimensional structure. This leads to impaired transport of bicarbonate and chloride across epithelial cell membranes, causing persistent bacterial infections and respiratory issues. Class III mutations result in a protein that reaches the cell surface but fails to react appropriately to cellular cues regulating channel gating. G551D is a well-known Class III mutation, where aspartic acid is substituted for glycine at position 551, resulting in a more closed state and lower channel open probability. These mutations can lead to serious respiratory issues and a lack of proper cellular function (Stanke and Tmmler, 2016).
PHENOTYPE EXPRESSIONS IN CYSTIC FIBROSIS
The genotype-phenotype correlation in Cystic Fibrosis (CF) is the link between specific CFTR mutations and the disease’s clinical symptoms. CF affects various organ systems, including the lungs, pancreas, liver, intestines and reproductive organs. The disease’s severity is influenced by specific CFTR mutations and environmental variables. Respiratory manifestations vary based on mutation type, with CF causing thick mucus production, chronic infections and lung inflammation (López-Valdez et al., 2021) as depicted in Figure 1. Chronic infections and inflammation are caused by genetic mutations, mucus accumulation and structural damage to the lungs. Infections from various bacteria are common in CF patients (Turcios, 2020).
The quality of life of patients with cystic fibrosis can be greatly affected by the severe Gastrointestinal (GI) symptoms of the disease. GI symptoms that are important in cystic fibrosis are listed in Table 1.
Cystic Fibrosis (CF) is a prevalent condition causing reproductive issues, particularly male infertility. The endocrine system can also be affected, leading to diabetes related to CFRD. The quality of life is significantly impacted by these systemic signs, necessitating comprehensive, multidisciplinary care. CF-affected females may experience thick cervical mucus, chronic lung infections, low nutritional status and diabetes. Advances in CF care have improved fertility and pregnancy outcomes. CF patients also face major metabolic and endocrine system disruptions, including insulin resistance, decreased insulin synthesis, poor nutritional status, stunted growth, vitamin D insufficiency, abnormal calcium metabolism and bone damage. Comprehensive management and regular monitoring are essential for improving the quality of life and long-term results (Jain et al., 2021).
EMERGING THERAPIES FOR CYSTIC FIBROSIS
CFTR Modulators
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulators are drugs that target the faulty CFTR protein to cure the underlying cause of Cystic Fibrosis (CF). The CFTR protein controls the flow of water and salt into and out of cells, particularly in the digestive and pulmonary systems. A accumulation of thick, sticky mucus caused by a defective CFTR protein can cause digestive and respiratory issues (Tice et al., 2021).
CFTR potentiators, which improve the CFTR protein’s cell surface function, have become a major focus of scientific research. These medications aid in the increased opening of CFTR channels, facilitating the more efficient passage of chloride ions. Clinically used CFTR potentiators include Ivacaftor (Kalydeco), which has proven effective in restoring CFTR channel activity (Csandy, Vergani, and Gadsby, 2019). Ivacaftor was the first CFTR modulator licensed by the U.S. Food and Drug Administration (FDA) in 2012 for the treatment of cystic fibrosis. Clinical trials have consistently shown that ivacaftor leads to significant improvements in lung function, reduced pulmonary exacerbations, weight gain, enhanced quality of life and fewer respiratory symptoms (Cho et al., 2019). Long-term studies confirm that ivacaftor provides sustained benefits, including prolonged improvements in lung function and lower rates of lung infections and hospitalizations over several years of use. Ivacaftor has revolutionized cystic fibrosis treatment for individuals with responsive mutations, offering significant clinical benefits and enhanced quality of life (Duckers et al., 2021).
CFTR correctors are pharmacological agents that improve the trafficking, folding and processing of the CFTR protein to the cell surface. They address mutations like F508del, preventing misfolding and proteolytic degradation. These medications increase functional protein at the cell surface and work synergistically with potentiators to enhance protein function. Lumacaftor, a drug approved by the FDA in 2015, is a combination therapy that improves the management of cystic fibrosis, particularly in patients with the F508del mutation in the CFTR gene (Cheng, Alexiou, and Rubenstein, 2019). It corrects misfolding of the CFTR protein, enhancing its flow to the cell surface and facilitating its movement to the cell surface. The combination therapy has shown significant improvements in lung function, pulmonary exacerbations and quality of life for patients with two copies of the F508del mutation. However, it also has potential side effects, including liver-related events, respiratory symptoms, elevated blood pressure, hypersensitivity reactions and drug interactions. Tezacaftor, a drug similar to lumacaftor, assists in the correct folding and trafficking of the CFTR protein to the cell surface. When paired with ivacaftor, tezacaftor is highly effective in treating cystic fibrosis patients with specific mutations, increasing lung function, decreasing pulmonary exacerbations and improving quality of life. The EVOLVE and EXPAND trials confirmed the drug combination’s effectiveness and safety, showing notable improvements for both heterozygous and homozygous F508del patients (Kuk and Taylor-Cousar, 2015).
CFTR amplifiers and readthrough agents are two pharmacological approaches aimed at improving CFTR function in Cystic Fibrosis (CF) patients. CFTR amplifiers increase the production of CFTR protein or enhance its stability at the cell surface, without directly correcting folding defects or enhancing channel gating (Dukovski et al., 2020). They work by boosting transcription or translation of CFTR mRNA, leading to increased CFTR protein production. VX-659, developed by Vertex Pharmaceuticals, is part of a combination therapy with tezacaftor and ivacaftor, marketed under the brand Trikafta. Readthrough agents overcome premature stop codons in the CFTR gene, enabling the production of a full-length, functional CFTR protein. Ataluren targets premature stop codons to facilitate full-length CFTR protein production (Dukovski et al., 2020).
Table 1:
Summary of GI manifestation in CF.
| Sl. No. | Condition | Description | Symptoms | References |
|---|---|---|---|---|
| 1 | Pancreatic insufficiency. | Pancreatic insufficiency, a condition where the pancreas is unable to produce enough digestive enzymes, affects the majority of CF patients. | Malnourishment, inability to thrive, steatorrhea (fatty feces) and inadequate nutrient absorption. | (Kunovsk; et al., 2021; Ritivoiu et al., 2023) |
| 2 | Meconium Ileus. | Meconium, or the first feces, is too thick and sticky to pass through the intestines in certain CF patients, a disease that exists from birth. | Vomiting, loose stools and abdominal distension soon after delivery. | (Donos et al., 2024; Tobias, Tillotson, Maloney, and Fialkowski, 2022) |
| 3 | Distal Intestinal Obstruction Syndrome (DIOS). | Occurs when the intestines are blocked by thicker intestinal contents. | Bloating, nausea, vomiting episodes and abdominal pain. | (Carroll, Green, and Gilchrist, 2021; Gilchrist, Green, and Carroll, 2021; Hort, Hameed, Middleton, and Pleass, 2020) |
| 4 | Gastroesophageal Reflux Disease (GERD). | Because of poor esophageal motility and increased abdominal pressure from coughing, GERD is common in people with cystic fibrosis. | Chest discomfort, heartburn and regurgitation. | (Bongiovanni et al., 2020) |
| 5 | Liver Disease | Because of bile duct blockage, Cystic Fibrosis (CF) can result in liver problems such as fatty liver, liver fibrosis and cirrhosis. | Increased liver enzymes, hepatomegaly (enlarged liver) and jaundice. | (Dana et al., 2022)(Staufer, 2020) |
| 6 | Nutritional Deficiencies. | CF patients frequently suffer shortages in other minerals including fat-soluble vitamins (A, D, E and K) as a result of malabsorption. | Poor immune function, anemia, bone disease and eyesight issues. | (Mailhot, Denis, Beauchamp-Parent, and Jomphe, 2023) |
| 7 | Small Bowel Bacterial Overgrowth (SIBO). | This happens when the small intestine’s general bacterial population experiences an unusual rise. | Bloating, discomfort in the abdomen and diarrhea. | (Efremova et al., 2023) |
| 8 | CF-related Diabetes (CFRD). | CFRD is a unique form of diabetes seen in CF patients due to damage to the insulin-producing cells in the pancreas. | Increased thirst, frequent urination, weight loss and fatigue. | (Frost, Walshaw, and Nazareth, 2022) |
Triple therapy, a combination of ivacaftor/tezacaftor/elexacaftor, is a treatment for Cystic Fibrosis (CF) patients with at least one Phe508del mutation in the CFTR gene. This treatment improves pulmonary function, reduces exacerbations and enhances quality of life. The CFTR modulators, such as elexacaftor, tezacaftor and ivacaftor, have shown significant clinical outcomes, including improved lung function, decreased exacerbations, improved quality of life and improved nutritional status (Konstan et al., 2020).
Gene Therapy Approaches
Gene Editing Techniques (e.g., CRISPR/Cas9)
CRISPR is a gene-editing technology developed by Streptococcus pyogenes that integrates foreign DNA into their genome. This technology involves the use of scaffolding trans-activating short RNAs (tracrRNAs) to interact with a nuclease protein, Cas9 (Konstan et al., 2020). However, the implementation of CRISPR-Cas9 technology is hampered by the unique physiology of Cystic Fibrosis (CF) lung. The efficiency of CRISPR-Cas9 depends on the target cell type, delivery method and genetic alterations site. Various editing methods, including prime, base and HITI editing, have their own advantages and drawbacks. HITI allows for precise insertion of genes without causing double-strand breaks, while base editing allows for direct conversion of DNA base pairs without double-strand breaks (Graham and Hart, 2021). Prime editing allows for precise genome editing without double-strand breaks, but efficiency may vary depending on the complexity of the edit and the target genomic region. Various techniques, such as electrophoration, nanoparticles and RNA-mediated delivery, have been used to deliver CRISPR-Cas9 to various cell types (Wei, Cheng, Min, Olson, and Siegwart, 2020).
Gene Replacement Strategies
Adenovirus (Ad) vectors are effective for gene delivery in cystic fibrosis patients, but require repeated administrations due to pro-inflammatory effects. Adeno-Associated Virus (AAV) vectors have longer in vivo persistence and potential for restoring CFTR function with minimal toxicity (Wang, Gessler, Zhan, Gallagher, and Gao, 2024). Non-viral vectors, which can carry larger DNA fragments, have lower efficacy and require repeated treatments due to transient gene expression, making them less effective for long-term lung function restoration in cystic fibrosis (Lee et al., 2021).
Other Therapeutic Strategies
Cystic Fibrosis (CF) is a lung disease that requires a multifaceted approach to manage. Anti-inflammatory agents, such as corticosteroids and NSAIDs, are used to reduce chronic inflammation in the lungs, which contributes to lung damage and disease progression. However, their use is limited by adverse effects and adsorption. Research into other anti-inflammatory strategies, such as specific antibodies and neutrophil-targeted therapies, is ongoing (Chmiel and Konstan, 2005). Mucociliary clearance enhancers help clear thick mucus from the lungs, improving respiratory function and reducing infection risk. Antimicrobial treatments, such as inhaled and systemic antibiotics, are crucial in managing CF by targeting chronic and recurrent respiratory infections. Long-term antibiotic therapy is often necessary to address antibiotic resistance and maintain lung function (Manos, 2021).
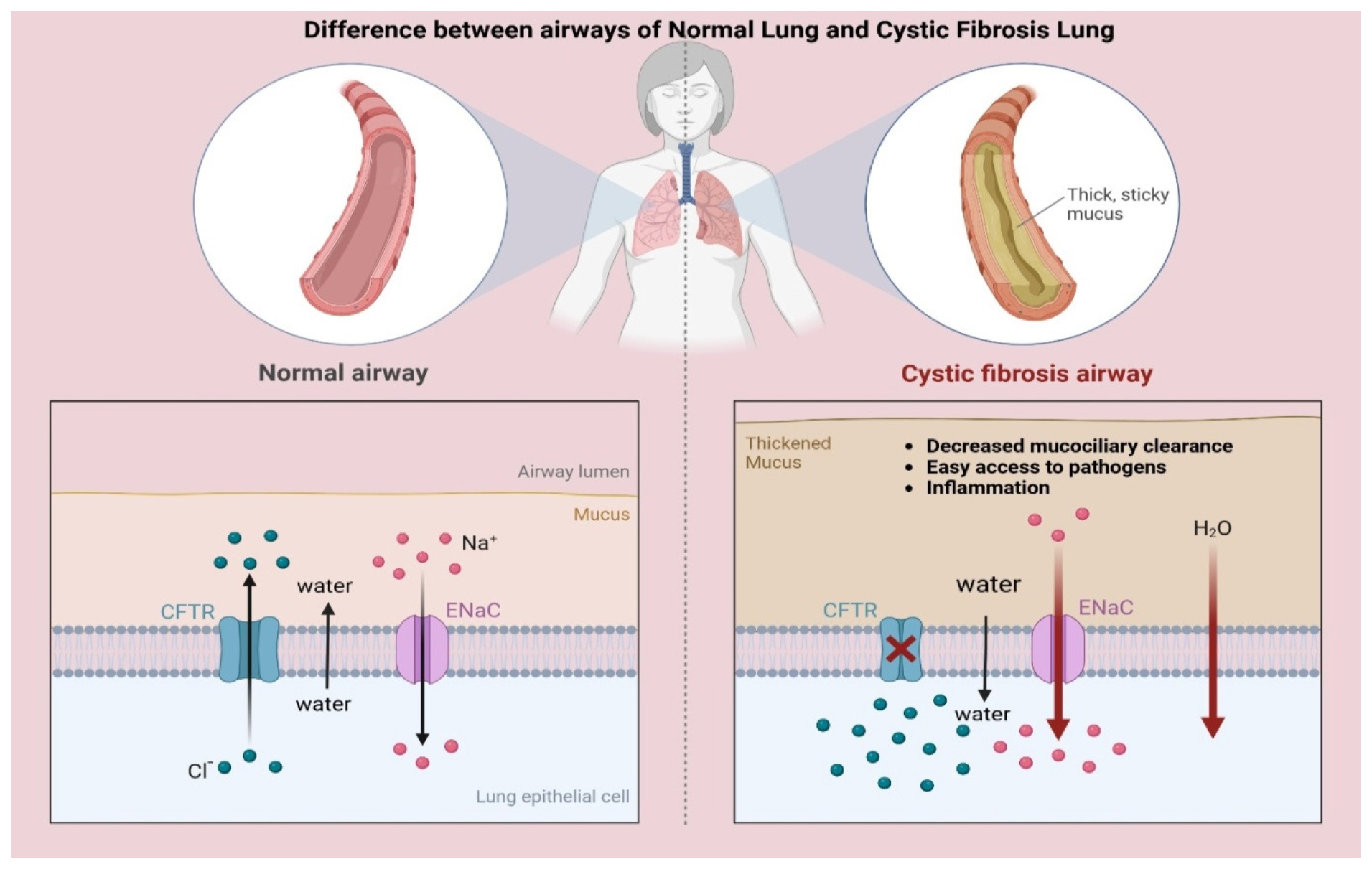
Figure 1:
Lung parenchymal changes in CF. Adapted from Cystic Fibrosis Airways by BioRender.com (2025). Retrieved from Retrieved from https://app.biorender.com/biorender-templates.
CLINICAL TRIALS AND RESEARCH
Ongoing clinical trials are essential for evaluating the effectiveness of these personalized therapies and understanding their long-term impacts on CF patients. Research into new models and delivery methods is also critical for advancing personalized medicine in CF.
A Phase 2B clinical trial evaluated the efficacy of a gene therapy for Cystic Fibrosis (CF) by delivering the therapy to patients’ airways over 12 months. This randomized, double-blind, placebo-controlled study found that the treatment significantly improved lung function. The main endpoint showed a 3.7% improvement, with a 95% confidence interval ranging from 0.1% to 7.3% and the result was statistically significant with a p-value of 0.031. Secondary endpoints, including Forced Vital Capacity (FVC) and CT gas trapping, also showed significant improvements with p-values of 0.031 and 0.048, respectively. Patients with higher baseline Forced Expiratory Volume (FEV1) experienced a notable 6.4% increase in FEV1 and those with milder disease also saw benefits, particularly in small airway disease markers like the Lung Clearance Index (LCI) (Alton et al., 2015; Alton et al., 2016).
A Phase II multicenter, double-blind, placebo-controlled study investigated the efficacy and safety of CFTR gene transfer using the tgAAVCF vector in patients with cystic fibrosis. The study focused on comparing the rate of recurrent sinusitis between tgAAVCF-treated and placebo-treated sinuses, using each patient as their own control. The results showed no significant differences in the levels of the pro-inflammatory cytokine IL-8 in both short-term (p=0.1155) and long-term (p=0.1653) analyses. However, there was a significant increase in the anti-inflammatory cytokine IL-10 over time (p=0.0261), suggesting that the delivery of the CFTR gene may have helped reduce inflammation in patients (Wagner et al., 2002).
The Roll-Over Study of Ivacaftor in children with cystic fibrosis under 6 years of age, who have a CFTR gating mutation, examined the effects of ivacaftor over 84 weeks with age- and weight-based dosing. The study found that ivacaftor was well tolerated in children aged 2 to 5, with sustained improvements in sweat chloride levels, weight, Body Mass Index (BMI) z-scores and fecal elastase-1 over the extended treatment period. However, 30% of the children developed elevated transaminase levels, indicating potential liver enzyme abnormalities. Despite this, the overall benefits of ivacaftor in this pediatric population were maintained throughout the study (Rosenfeld et al., 2019).
CURRENT CHALLENGES
Despite advancements in cystic fibrosis treatment, there are still issues that affect patients’ quality of life and treatment effectiveness. Current treatments focus on symptom management rather than treating the underlying genetic issue, leading to thick, sticky mucus. CFTR modulators have limited efficacy, with 10% of patients not benefiting. High costs make them unaffordable, particularly in low- and middle-income countries. Antibiotic resistance is another major issue and lengthy treatment plans affect patients’ quality of life and adherence to treatment (Burgener and Moss, 2018; Altabee et al., 2022).
FUTURE PERSPECTIVE
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) treatment is promising due to advancements in gene editing, medication and personalized care. CFTR modulators, like ivacaftor, lumacaftor and elexacaftor, have improved lung function and quality of life for many patients. Research aims to develop next-generation modulators with increased efficacy. Gene editing technologies, like CRISPR-Cas9, offer a long-term cure by directly repairing the faulty CFTR gene in patient cells. Clinical trials are ongoing to investigate the safety and effectiveness of these strategies. Stem cell therapy is also being explored to repair healthy lung tissue. Personalized medicine, which uses patient-specific data to customize medications, is a growing trend in medicine. Combining these approaches could significantly improve cystic fibrosis patients’ quality of life and shift the focus of CFTR therapy from symptom relief to a permanent cure. Collaboration between researchers, physicians and the pharmaceutical industry is crucial for advancing these treatments (Abdelnour, Xie, Hassanin, Zuo, and Lu, 2021; Kramer and Clancy, 2016).
CONCLUSION
Despite significant advancements in cystic fibrosis treatment through CFTR modulators, major challenges remain. Current therapies are non-responsive to many mutations, leaving a significant portion of the CF population without effective treatment options. Genetic therapy faces practical obstacles like high costs, efficiency issues and risk of off-target effects. Limited long-term safety and efficacy data, delivery difficulties and ethical concerns complicate its application. To overcome these limitations, next-generation gene modulators must target non-responsive mutations, stem cell therapy and one-time treatment options like genetic modifications.
ACKNOWLEDGEMENT
I would like to express my sincere gratitude to the research community whose invaluable contributions on cystic fibrosis cited for this review article.
CONFLICT OF INTEREST
The author declares that there is no conflict of interest.
ABBREVIATIONS
| CF | Cystic fibrosis |
|---|---|
| CFTR | Cystic Fibrosis Transmembrane Regulator |
| US | United States |
| RNA | Ribonucleic acid |
| TMD | Transmembrane domain |
| NBD | Nucleotide-binding domain |
| RD | Regulatory domain |
| GI | Gastrointestinal |
| CFRD | Cystic Fibrosis related diabetes |
| FDA | Food and Drug Administration |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| HITI | Homology-independent targeted integration |
| DNA | Deoxyribonucleic acid |
| AAV | Adeno-Associated Virus |
| NSAIDs | Non-Steroidal Anti-inflammatory Drugs |
| FVC | Forced vital capacity |
| CT | Computed tomography |
| FEV | Forced expiratory volume |
| LCI | Lung clearance index |
| tgAAVCF | adeno-associated cystic fibrosis transmembrane conductance regulator (CFTR) viral vector/gene construct |
| BMI | Body Mass Index |
REFERENCES
- Abdelnour, S. A., Xie, L., Hassanin, A. A., Zuo, E., & Lu, Y. (2021). The potential of CRISPR/ Cas9 gene editing as a treatment strategy for inherited diseases. Frontiers in Cell and Developmental Biology, 9, Article 699597. CrossRef]
- Altabee, R., Carr, S. B., Abbott, J., Cameron, R., Office, D., Matthews, J., Simmonds, N., Cosgriff, R., Turner, D., & Whitty, J. (2022). Exploring the nature of perceived treatment burden: A study to compare treatment burden measures in adults with cystic fibrosis [version 1; peer review: 2 approved]. NIHR Open Research, 2, 36. CrossRef]
- Alton, E. W. F. W., Armstrong, D. K., Ashby, D., Bayfield, K. J., Bilton, D., Bloomfield, E. V., Waller, M. D. (2015). Cystic fibrosis gene therapy clinical trial demonstrates beneficial effect on lung function, 2600(15), 1-9. CrossRef]
- Alton, E. W. F. W., Armstrong, D. K., Ashby, D., Bayfield, K. J., Bilton, D., Bloomfield, E. V., Boyd, A. C., Brand, J., Buchan, R., Calcedo, R., Carvelli, P., Chan, M., Cheng, S. H., Collie, D. S., Cunningham, S., Davidson, H. E., Davies, G., Davies, J. C., Davies, L. A., Wolstenholme-Hogg, P. (2016). A randomised, double-blind, placebo-controlled trial of repeated nebulisation of non-viral cystic fibrosis transmembrane conductance regulator (CFTR) gene therapy in patients with cystic fibrosis. Efficacy and Mechanism Evaluation, 3(5), 1-210. CrossRef]
- Bongiovanni, A., Manti, S., Parisi, G. F., Papale, M., Mulè, E., Rotolo, N., & Leonardi, S. (2020). Focus on gastroesophageal reflux disease in patients with cystic fibrosis. World Journal of Gastroenterology, 26(41), 6322-6334. CrossRef]
- Burgener, E. B., & Moss, R. B. (2018). Cystic fibrosis transmembrane conductance regulator modulators: Precision medicine in cystic fibrosis. Current Opinion in Pediatrics, 30(3), 372-377. CrossRef]
- Carroll, W., Green, J., & Gilchrist, F. J. (2021). Interventions for preventing distal intestinal obstruction syndrome (DIOS) in cystic fibrosis. The Cochrane Database of Systematic Reviews, 12(12), Article CD012619. CrossRef]
- Cheng, P. C., Alexiou, S., & Rubenstein, R. C. (2019). Safety and efficacy of treatment with lumacaftor in combination with ivacaftor in younger patients with cystic fibrosis. Expert Review of Respiratory Medicine, 13(5), 417-423. CrossRef]
- Chmiel, J. F., & Konstan, M. W. (2005). Anti-inflammatory medications for cystic fibrosis lung disease: Selecting the most appropriate agent. Treatments in Respiratory Medicine, 4(4), 255-273. CrossRef]
- Cho, D.-Y., Zhang, S., Lazrak, A., Grayson, J. W., Peña Garcia, J. A., Skinner, D. F., Lim, D. J., Mackey, C., Banks, C., Matalon, S., & Woodworth, B. A. (2019). Resveratrol and ivacaftor are additive G551D CFTR-channel potentiators: Therapeutic implications for cystic fibrosis sinus disease. International Forum of Allergy and Rhinology, 9(1), 100-105. CrossRef]
- Csanády, L., Vergani, P., & Gadsby, D. C. (2019). STRUCTURE, GATING and REGULATION OF THE CFTR ANION CHANNEL. Physiological Reviews, 99(1), 707-738. CrossRef]
- Dana, J., Debray, D., Beaufrère, A., Hillaire, S., Fabre, M., Reinhold, C., Baumert, T. F., Berteloot, L., & Vilgrain, V. (2022). Cystic fibrosis-related liver disease: Clinical presentations, diagnostic and monitoring approaches in the era of CFTR modulator therapies. Journal of Hepatology, 76(2), 420-434. CrossRef]
- Donos, M. A., Ghiga, G., Trandafir, L. M., Cojocaru, E., Ţarcă, V., Butnariu, L. I., Bernic, V., Moroşan, E., Roca, I. C., Mîndru, D. E., & Ţarcă, E. (2024). Diagnosis and management of simple and complicated meconium ileus in cystic fibrosis, a systematic review. Diagnostics, 14(11), 1179. CrossRef]
- Duckers, J., Lesher, B., Thorat, T., Lucas, E., McGarry, L. J., Chandarana, K., & De Iorio, F. (2021). Real-world outcomes of ivacaftor treatment in people with cystic fibrosis: A systematic review. Journal of Clinical Medicine, 10(7), Article 1527. CrossRef]
- Dukovski, D., Villella, A., Bastos, C., King, R., Finley, D., Kelly, J. W., Morimoto, R. I., Hartl, F. U., Munoz, B., Lee, P.-S., Zecevic, M., & Miller, J. P. (2020). Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society, 19(5), 733-741. CrossRef]
- Efremova, I., Maslennikov, R., Poluektova, E., Vasilieva, E., Zharikov, Y., Suslov, A., Letyagina, Y., Kozlov, E., Levshina, A., & Ivashkin, V. (2023). Epidemiology of small intestinal bacterial overgrowth. World Journal of Gastroenterology, 29(22), 3400-3421. CrossRef]
- Frost, F., Walshaw, M. J., & Nazareth, D. (2022). Cystic fibrosis-related diabetes: An update. QJM: An International Journal of Medicine, 115(9), 582-585. CrossRef]
- Gilchrist, F. J., Green, J., & Carroll, W. (2021). Interventions for treating distal intestinal obstruction syndrome (DIOS) in cystic fibrosis. The Cochrane Database of Systematic Reviews, 12(12), Article CD012798. CrossRef]
- Graham, C., & Hart, S. (2021). CRISPR/Cas9 gene editing therapies for cystic fibrosis. Expert Opinion on Biological Therapy, 21(6), 767-780. CrossRef]
- Hort, A., Hameed, A., Middleton, P. G., & Pleass, H. C. (2020). Distal intestinal obstruction syndrome: An important differential diagnosis for abdominal pain in patients with cystic fibrosis. ANZ Journal of Surgery, 90(5), 681-686. CrossRef]
- Jain, R., Kazmerski, T. M., Aitken, M. L., West, N., Wilson, A., Bozkanat, K. M., Montemayor, K., von Berg, K., Sjoberg, J., Poranski, M., & Taylor-Cousar, J. L. (2021). Challenges faced by women with cystic fibrosis. Clinics in Chest Medicine, 42(3), 517-530. CrossRef]
- Konstan, M. W., VanDevanter, D. R., Rowe, S. M., Wilschanski, M., Kerem, E., Sermet-Gaudelus, I., DiMango, E., Melotti, P., McIntosh, J., De Boeck, K., & ACT CF Study Group. (2020). Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled ataluren Confirmatory Trial in cystic fibrosis (ACT CF). Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society, 19(4), 595-601. CrossRef]
- Kramer, E. L., & Clancy, J. P. (2016). CFTR modulator therapies in pediatric cystic fibrosis: Focus on ivacaftor. Expert Opinion on Orphan Drugs, 4(10), 1033-1042. CrossRef]
- Kuk, K., & Taylor-Cousar, J. L. (2015). Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: Current evidence and future prospects. Therapeutic Advances in Respiratory Disease, 9(6), 313-326. CrossRef]
- Kunovský, L., Dít?, P., Jabandžiev, P., Eid, M., Poredská, K., Vaculová, J., Sochorová, D., Janeček, P., Tesaříková, P., Blaho, M., Trna, J., Hlavsa, J., & Kala, Z. (2021). Causes of exocrine pancreatic insufficiency other than chronic pancreatitis. Journal of Clinical Medicine, 10(24), 5779. CrossRef]
- Lee, J.-A., Cho, A., Huang, E. N., Xu, Y., Quach, H., Hu, J., & Wong, A. P. (2021). Gene therapy for cystic fibrosis: New tools for precision medicine. Journal of Translational Medicine, 19(1), 452. CrossRef]
- Liu, F., Zhang, Z., Levit, A., Levring, J., Touhara, K. K., Shoichet, B. K., & Chen, J. (2019). Structural identification of a hotspot on CFTR for potentiation. Science, 364(6446), 1184-1188. CrossRef]
- López-Valdez, J. A., Aguilar-Alonso, L. A., Gándara-Quezada, V., Ruiz-Rico, G. E., Ávila-Soledad, J. M., Reyes, A. A., & Pedroza-Jiménez, F. D. (2021). Cystic fibrosis: Current concepts. Boletín Médico del Hospital Infantil de México, 78(6), 584-596. htt ps://doi.org/10.24875/BMHIM.20000372
- Mailhot, G., Denis, M.-H., Beauchamp-Parent, C., & Jomphe, V. (2023). Nutritional management of people living with cystic fibrosis throughout life and disease continuum: Changing times, new challenges. Journal of Human Nutrition and Dietetics, 36(5), 1675-1691. CrossRef]
- Manos, J. (2021). Current and emerging therapies to combat cystic fibrosis lung infections. Microorganisms, 9(9), Article 1874. CrossRef]
- Pereira, S. V.-N., Ribeiro, J. D., Ribeiro, A. F., Bertuzzo, C. S., & Marson, F. A. L. (2019). Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools. Scientific Reports, 9(1), 6234. CrossRef]
- Rafeeq, M. M., & Murad, H. A. S. (2017). Cystic fibrosis: Current therapeutic targets and future approaches. Journal of Translational Medicine, 15(1), 84. CrossRef]
- Ritivoiu, M.-E., Drăgoi, C. M., Matei, D., Stan, I. V., Nicolae, A. C., Craiu, M., Dumitrescu, I.- B., & Ciolpan, A. A. (2023). Current and future therapeutic approaches of exocrine pancreatic insufficiency in children with cystic fibrosis in the era of personalized medicine. Pharmaceutics, 15(1), 162. CrossRef]
- Rosenfeld, M., Cunningham, S., Harris, W. T., Lapey, A., Regelmann, W. E., Sawicki, G. S., Southern, K. W., Chilvers, M., Higgins, M., Tian, S., Cooke, J., Davies, J. C., & KLIMB study group. (2019). An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2-5 years (KLIMB). Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society, 18(6), 838-843. CrossRef]
- Stanke, F., & Tümmler, B. (2016). Classification of CFTR mutation classes. The Lancet. Respiratory Medicine, 4(8), e36. CrossRef]
- Staufer, K. (2020). Current treatment options for cystic fibrosis-related liver disease. International Journal of Molecular Sciences, 21(22), 8586. CrossRef]
- Sui, H., Xu, X., Su, Y., Gong, Z., Yao, M., Liu, X., Zhang, T., Jiang, Z., Bai, T., Wang, J., Zhang, J., Xu, C., & Luo, M. (2022). Gene therapy for cystic fibrosis: Challenges and prospects. Frontiers in Pharmacology, 13, Article 1015926. CrossRef]
- Tice, J. A., Kuntz, K. M., Wherry, K., Seidner, M., Rind, D. M., & Pearson, S. D. (2021). The effectiveness and value of novel treatments for cystic fibrosis. Journal of Managed Care and Specialty Pharmacy, 27(2), 276-280. CrossRef]
- Tobias, J., Tillotson, M., Maloney, L., & Fialkowski, E. (2022). Meconium ileus, distal intestinal obstruction syndrome and other gastrointestinal pathology in the cystic fibrosis patient. The Surgical Clinics of North America, 102(5), 873-882. CrossRef]
- Turcios, N. L. (2020). Cystic fibrosis lung disease: An overview. Respiratory Care, 65(2), 233-251. CrossRef]
- Wagner, J. A., Nepomuceno, I. B., Messner, A. H., Moran, M. L., Batson, E. P., Dimiceli, S., Brown, B. W., Desch, J. K., Norbash, A. M., Conrad, C. K., Guggino, W. B., Flotte, T. R., Wine, J. J., Carter, B. J., Reynolds, T. C., Moss, R. B., & Gardner, P. (2002). A Phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Human Gene Therapy, 13(11), 1349-1359. CrossRef]
- Wang, J.-H., Gessler, D. J., Zhan, W., Gallagher, T. L., & Gao, G. (2024). Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduction and Targeted Therapy, 9(1), 78. CrossRef]
- Wei, T., Cheng, Q., Min, Y.-L., Olson, E. N., & Siegwart, D. J. (2020). Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nature Communications, 11(1), 3232. CrossRef]
- Zhang, Z., Liu, F., & Chen, J. (2017). Conformational changes of CFTR upon phosphorylation and ATP binding. Cell, 170(3), 483-491.e8. CrossRef]