ABSTRACT
Background: In the management of bleeding disorders, a synthetic lysine analogue Tranexamic acid is one of the drugs commonly used. Synthetic lysine analogue used in the management of bleeding disorders. The main purpose of the research aimed at developing and validating the new, simple, and cost–effective solid phase extraction method for evaluating tranexamic acid (TX) in human plasma by Ultra–performance liquid chromatography–Mass spectrometry (UPLC–MS/MS) using tranexamic acid as Internal Standard. Materials and Methods: Excellent separation and elution was fulfilled using acetonitrile: 100mM ammonium formate pH 3.5(60:40 v/v) as the mobile phase, at a flow rate of 0.300 mL/min with the injection volume of 5µL. Chromatographic analysis of the analyte and IS was initiated under isocratic conditions with an aim to develop a simple separation process with a short run time. Results: The method showed a calibration curve range from 150.00ng/ml- 15,004.00ng/ml. validation of method was determined by using various supporting data of precision and accuracy with percentage recovery of 76.01% and 78.61% for TX and TXD2 respectively. Conclusion: An easy and sensitive UPLC–MS/MS method has been developed for the evaluation of TX in human plasma. This developed method provides excellent linearity over a wide range of TX, sensitive, and precise. Hence this technique is ideal and suitable for clinical pharmacokinetic studies of tranexamic acid.
INTRODUCTION
Tranexamic acid (TX) is chemically Trans-4- amino methyl– cyclo hexane carboxylic acid it is a synthetic lysine analogue used as antifibrinolytic agent which acts by inhibition of lysine binding at plasminogen receptors.1–5 Due to the recent development on phosphorlipid–induced matrix effects during human plasma analysis of tranexamic acid, our aim was to develop a appropriate and reliable method, and its validation for the determination of tranexamic acid from human plasma by UPLC–Mass Spectrometry/ Mass Spectrometry (UPLC–MS/MS). Based on the literature review it was found that there are many methods such as LC, UV, HPLC, LC–MS/MS, HPLC–UPLC, Fluorimetry, and gas chromatography electron capture detector (GC–ECD) are available for the determination of tranexamic acid from human plasma.6–9 However, they need either pre-column, or post-column derivatization which is complex, and time consuming.10 Thus they are not suitable for analysis of large numbers of samples. In addition, derivatization may introduce large assay variation. To overcome this issues a newer method have been developed based on LC–MS/MS and UPLC–MS/MS.8,11
Liquid Chromatography/Mass Spectrometry (LC/MS) is the hyphenated technique and a prevailing analytical method that combines the resolving power of liquid chromatography with the detection specificity of mass spectrometry. Liquid chromatography separates the sample components followed by introduction to the mass spectrometer (MS). The MS creates and detects charged ions.11
The objective of this research work was (1) To develop a simple, selective, sensitive, and reproducible bioanalytical method for estimation of tranexamic acid in human plasma by UPLC–Mass Spectrometry. (2) To achieve good separation and rapid elution of tranexamic acid (3) To ensure reliability, as human plasma contains many chemical constituents, and other materials which may interact with the estimation of tranexamic acid, it is necessary to develop an ideal method to estimate the tranexamic acid in human plasma by minimising the interaction of components of human plasma.12 Hence this UPLC–Mass chromatography method is developed for the estimation of tranexamic acid in human plasma which would be more accurate and useful in pharmacological disorders like bleeding control.
MATERIALS AND METHODS
Materials
Tranexamic acid (TX), Reference Standard, and Tranexamic acid D2 (TXD2), as Internal Standard (IS) were purchased from Clear synth Labs Limited, Mumbai, India. Methanol and Acetonitrile (HPLC/LCMS grade) from Honey well research chemicals. Ammonium formate and formic acid were purchased from Merck. Human plasma was obtained as gift sample from Symbiosis, Ahmedabad, India. Ultra–pure water was obtained from Novitium Pharma Private Limited, Chennai, India. All other chemicals used in this research were of analytical grade.
Methods
Preparation of standard solution
Tranexamic acid (TX)
Tranexamic acid was weighed and dissolved in a 20 mL volumetric flask with water and made up to required volume with water to obtain an equivalent concentration of 2000 microgram/ml and labelled as TX–MSX. Suitable dilutions were made to achieve final concentrations of 150–15,000ng /ml.13
Method development and method validation Preparation of sample solution
Added 50 µL of TXIS–I (Appx. 2500 ng/mL) into labelled polypropylene tubes/ria vials. 100 µL of plasma sample was pipetted out to respectively labelled polypropylene tubes/ ria vials and vortexed. 500 µL of 20% Formic acid in water was added to each tube and vortexed. The SPE cartridge (Strata–X-C 33 µm, 30 mg/mL) with 0.500 mL of methanol and centrifuged at 2000rpm for 1 min at 10±3 °C followed by 0.500 mL of 2% formic acid in water and centrifuged at 2000rpm for 1 min at 10±3 °C. The supernatant was loaded onto the SPE cartridge and centrifuged at 2000rpm for 2min at 10±3 °C. or With the SPE cartridge as follows.1 mL of 2% formic acid in water and centrifuge at 2000 rpm for 1min at 10±3 °C, followed by 1 mL of methanol, and centrifuge at 2000rpm for 2min at 10±3 °C. Eluted the SPE cartridge with 300 µL of Elution solvent and centrifuged at 2000rpm for 2 min at 10±3 °C for twice in to labelled polypropylene tube / ria vials. The eluent sample was evaporated to dryness at 40 °C ± 3 °C in LV evaporator. The dried residue was reconstituted with 500 µL of reconstitution solution and vortexed. The sample was transferred into auto sampler vials/ polypropylene shell vials and cap for injection.
UPLC–MS/MS analysis
Using Water Acquity system (Massachusetts, US) equipped with binary solvent delivery pump, tunable mass detector, and an auto sampler UPLC was performed. The Mass spectrometer used Waters XEVO Triple Quadruple Detector (TQD) along with Electro spray Ionisation. The chromatographic separation was done by suitable mobile and stationary phase with required injection volume and flow rate. Here Electro spray Ionisation source was used for the ionisation of polar solvent and it has a very good catalyst systems. The data system Mass lynx Version 4.1 software had been used for the quantification purpose.8,12,13
Chromatographic conditions
The analytical column used for the study was synergic C18 X 2.5 µm, polar–RP 100Å (100×3.0 mm) and the column maintained at the temperature of 40 °C. The mobile phase composition used was acetonitrile and 100mM solution of ammonium formate in the ratio of (60:40) respectively with the pH 3.5. The flow rate at which the mobile phase flows was set to be at 0.300 mL/min. In the auto sampler, the temperature was set to be 5 °C and the injection volume was set at 5µL. The run time was 3.00 min.
Validation of analytical method
RESULTS
Standard calibration curve
The Calibration curve was found to be linear. The slope was found to be 0.000,289,681 and the intercept was 0.0,116,366 and the determination coefficients (R2) were 0.997,440. These results indicate ample reliability and reproducibility of this method within the analytical range from 150–15,004ng/mL. The representative chromatograms are shown in Figure 1 and the values are shown in Table 1.
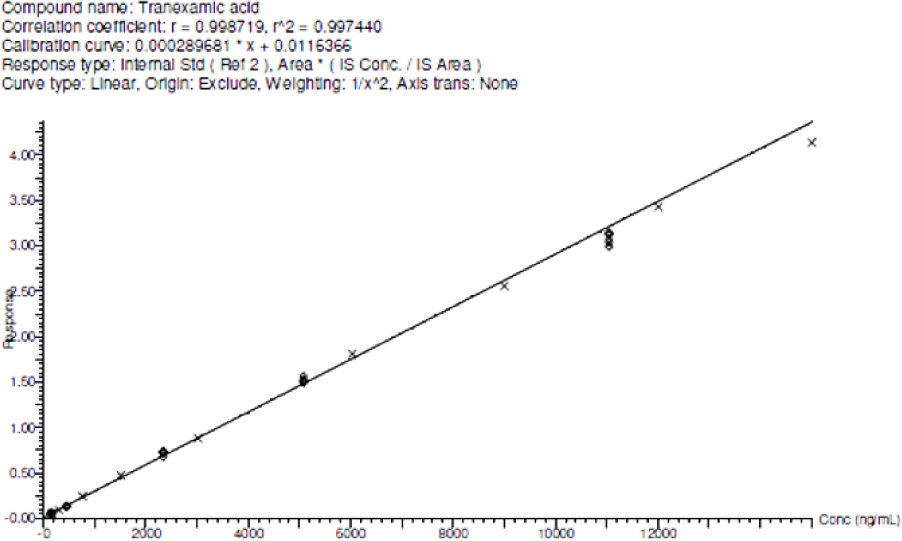
Figure 1.
Calibration curve for Tranexamic acid.
| Concentration ofCC | Mean | S.D | %CV | % Nominal | % Deviation |
|---|---|---|---|---|---|
| STD 1150.00 (ng/mL) | 149.44 | 6.56 | 4.39 | 99.06 | -0.94 |
| STD 2301.00 (ng/mL) | 298.60 | 24.30 | 8.14 | 99.00 | -1.00 |
| STD 3753.00 (ng/mL) | 787.72 | 40.00 | 5.08 | 104.47 | 4.47 |
| STD 41,507.00 (ng/mL) | 1582.12 | 23.71 | 1.5 | 104.92 | 4.92 |
| STD 53,015.00 (ng/mL) | 3,121.04 | 64.30 | 2.06 | 103.48 | 3.48 |
| STD 66,031.00 (ng/mL) | 6,090.91 | 97.87 | 1.61 | 100.98 | 0.98 |
| STD7 9,002.00 (ng/mL) | 8,744.70 | 43.26 | 0.49 | 97.13 | -2.87 |
| STD 812,003.00 (ng/mL) | 11,548.98 | 260.06 | 2.25 | 96.21 | -3.79 |
| STD 915,004.00 (ng/mL) | 14,216.23 | 301.05 | 2.12 | 94.74 | -5.26 |
Acceptance Criteria
Mean accuracy (% Nominal) must be within ± 15.0 % of nominal value for calibration standards except LLOQ standard (i.e. STD 1) for which it must be within ± 20.0 % of nominal value.
Precision (% CV) ≤ 15.0 % for each calibration standards except LLOQ standard (i.e. STD 1) for which it must be ≤ 20.0 %. The representative chromatograms are shown in Figure 2(a)–2(c) and the values are shown in Table 2.
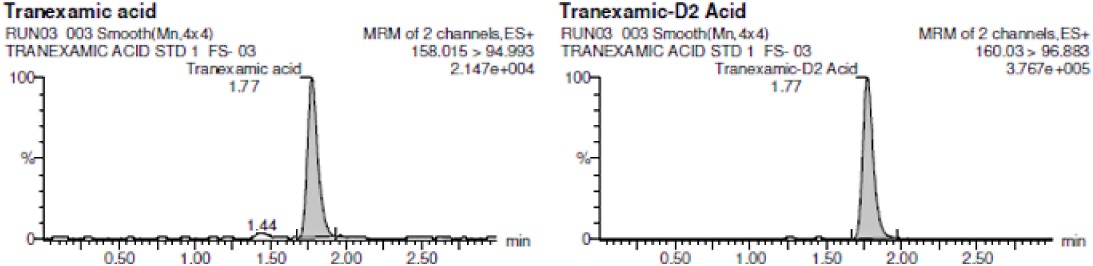
Figure 2 A:
Representative chromatogram of blank human plasma spiked with internal standard.

Figure 2 B:
Representative Chromatogram of LLOQ level.
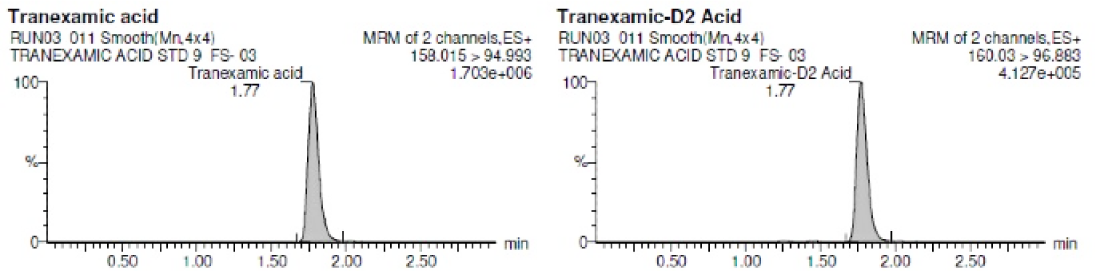
Figure 2 C:
Representative Chromatogram of ULOQ level.
| Type of assay | Parameters | LLOQQC 151.00 ng/Ml |
LQC 448.00 ng/mL |
MQC 505.00 ng/Ml |
HQC 11,043.00 ng/mL |
|---|---|---|---|---|---|
| Inter-assay P&A (n=18) | Inter run mean | 160.30 | 450.77 | 4,961.85 | 10,281.79 |
| Inter run SD | 14.79 | 32.27 | 163.06 | 470.99 | |
| Inter run CV% | 9.23 | 7.16 | 3.29 | 4.58 | |
| Inter run % Nominal | 106.16 | 100.62 | 98.25 | 93.11 | |
| Inter run % deviation | 6.16 | 0.62 | -1.75 | -6.89 |
Acceptance Criteria
QCs Inter-Run (from acceptable runs only) (Between runs): Mean accuracy must be within ± 20.0 % for LLOQQC and within ± 15.0 % of nominal value for other QCs. Precision ≤ 20.0 % for LLOQQC and ≤ 15.0 % for other QCs.
Method Development and its Validation
Parameters in mass spectrometry like fragmentation pattern and ionisation mode are the main task in tuning to achieve respective fragmented ions and response for both TX and TXD2 which were shown in Figure 3(a) and Figure 3(b).
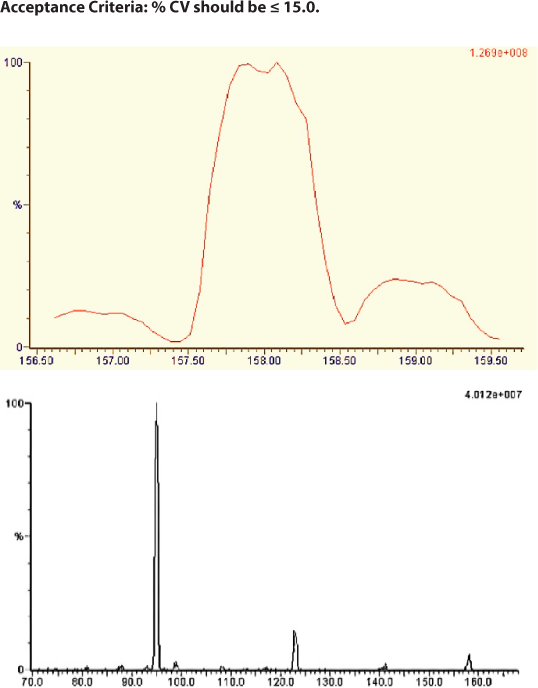
Figure 3.
Mass spectrum Tranexamic acid daughter ions.
The mass spectroscopy instrumental parameters were optimised. The ionisation mode was positive ion electrospray, the source temperature was 150 °C. The voltage of capillary, desolvation gas flow, cone gas flow, and probe flow rate is 2.5 kV, 1000 L/hr, 50 L/hr, 300μl/min respectively. For both TX and TXD2 the dwell time was 0.100 sec. The collisionally associated dissociation (CAD) mass spectrum of TX shows production of characteristic product ions at m/z 141.00, 122.97, 98.83 and 94.99. The main product ion at m/z was 94.99 for TX.
Limit of Quantification (LOQ)
The value of LOQ signal–to-noise (S/N) was found for 6 injections of HC. The mean value is 97.99 which are in the acceptance range S/N ≥ 5:1. The values are shown in Table 3–5.
| Sample level | Recovery | Mean | SD | %CV |
|---|---|---|---|---|
| HQC | 73.28 | 76.0100 | 3.51,051 | 4.62 |
| MQC | 74.78 | |||
| LQC | 79.97 |
| Sample level | Recovery | Mean | SD | %CV |
|---|---|---|---|---|
| HQC | 77.74 | 78.6,067 | 1.05,401 | 1.34 |
| MQC | 79.78 | |||
| LQC | 78.30 |
| Actual concentration (μg/mL) | 22,507.000 | 22,507.000 |
|---|---|---|
| Dilution factor | 2 | 4 |
| Mean | 20,944.84 | 22,191.30 |
| SD | 729.44 | 695.20 |
| %CV | 3.48 | 3.13 |
| %Nominal | 93.06 | 98.60 |
Acceptance Criteria
Accuracy should be within ± 15.0 % of nominal value, at least 4 out of 6 dilution QC samples must be within specifications. Mean Accuracy of QCs at each level should be within ± 15.0 %.
Precision (CV) ≤ 15.0
Stability studies
In freeze thaw stability over 5 cycles, it was found that no significant change was observed in concentration of TX in matrix, Neat standard stability for 8 days at REF condition, and Stability of Tranexamic acid for 24.5 hrs at RT and 61.0 hrs at auto sampler.14 The values are shown in Table 6–9.
| Sl. No. | QC concentration | Mean | SD | %CV | %Nominal | %Deviation |
|---|---|---|---|---|---|---|
| 1 | LOW(448.00 ng/mL) | 428.02 | 14.13 | 3.30 | 95.54 | -4.46 |
| 2 | HIGH(11,043.00 ng/mL) | 10,719.22 | 442.91 | 4.13 | 97.07 | -2.93 |
| Against Re injected calibration curve | ||||||
|---|---|---|---|---|---|---|
| Sl. No. | QC concentration | Mean | SD | %CV | %Nominal | %Deviation |
| 1 | LOW(448.00 ng/mL) | 422.38 | 6.88 | 1.63 | 94.28 | -5.72 |
| 2 | HIGH(11,043.00 ng/mL) | 10,709.33 | 267.32 | 2.50 | 96.98 | -3.02 |
| Mean | SD | %CV | Mean test/ Mean Ref (%) | ||
|---|---|---|---|---|---|
| TX | TX -NS–TEST | 113.735 | 268.15 | 0.24 | 100.48 |
| TX -NS–REF | 113.192 | 572.84 | 0.51 | ||
| TX IS | TX -NS–TEST | 77.093 | 364.32 | 0.47 | 100.38 |
| TX -NS–REF | 76.801 | 138.01 | 0.18 |
| Analyte spiking solution | Mean | SD | %CV | Mean test/ Mean Ref (%) |
|---|---|---|---|---|
| TX–ASS-TEST | 112.009 | 3.307.85 | 2.95 | 99.72 |
| TX–ASS-REF | 112.323 | 863.38 | 0.77 |
Acceptance criteria
Accuracy =% Nominal for QC at each concentration should be within 85–115 (± 15.0 %), at least 4 out of 6 individual concentrations must be within specification. Precision= % Deviation must be ≤ 15% for each concentration tested.
DISCUSSION
The main objective of this research activity was the development and validation of bio analytical method for the estimation of tranexamic acid. The developed method was simple, selective, sensitive, rapid, and reproducible for the quantification of tranexamic acid from plasma samples. The developed bioanalytical method was performed as per the FDA guidelines. UPLC with mass detector was selected for this study; Chromatographic conditions and extraction procedures were optimised to achieve the desirable results. The R2 value of the calibration curve of the analyte was 0.997,440 and all the parameters were performed by standard acceptance criteria. The mass spectrometric parameters optimization was performed in positive mode of ionisation due to its higher intensity by direct injection of neat solutions of both TX and TXIS. Generally, the estimation of tranexamic acid in any formulation or drug delivery system would be effective and accurate due to its high concentration and free from interaction between the tranexamic acid, and other excipients used in the formulation. But in human plasma, the concentration of tranexamic acid would be in low level which creates challenges in its estimation. So the method developed should be sensitive for low levels of tranexamic acid. Other parameters, such as composition, and nature of the mobile phase, selection of column were optimised through several trials to achieve best resolution. Separation of TX was carried out under various combinations of mobile phase with different columns. A good separation and rapid elution (run time 3 min and the analyte was eluted at 1.7min) were achieved using methanol and 0.1% formic acid with the flow rate of 0.300 ml/min and the injection volume is 5µl. Here, the solid–phase extraction method was selected as optimised method for TX, and TXIS from the plasma samples. Under isocratic conditions, chromatographic evaluation of the analyte, and IS were performed in order to provide a simple separation method with a brief run time. With several sources of information about batch precision and accuracy, a method validation has been established, with recovery percentages of 76.01% and 78.61% for TX and TXIS, respectively. The developed method was subjected for various stability studies such as freeze thaw stability with 5 cycle and bench top stability at 17.5 hrs at room temperature, dry extract stability at 28.0 hrs at room temperature, processed sample stability at room temperature, and 78 hrs at auto sampler, the whole blood stability at 2.0 hrs for both the condition of room temperature and ice bath. The analyte spiking solution stability is proven for 10 days at refrigerator condition and for 6.5 hrs at room temperature. The master stock stability of analyte and internal standard has proven for 10 days at refrigerator condition. The present study data obtained from this research would be a useful tool in the clinical pharmacokinetic determination of TX in human plasma samples.14–16
CONCLUSION
This UPLC–MS/MS method is a simple, rapid, specific and reproducible method for the determination of Tranexamic acid present in human plasma sample and it was developed, and validated as per FDA guidelines. The obtained all results was showed the under–acceptance limit only. This method needs a small quantity of the sample and good extraction recovery from the plasma and a readily available as an internal standard. Therefore, the proposed method can be used for monitoring the plasma concentration of Tranexamic acid present in patient samples by UPLC–MS/MS.
References
- Array. Martindale: The complete drug reference. 2009;36:1064-82. [Google Scholar]
- Cai J, Ribkoff J, Olson S, Raghunathan V, Al-Samkari H, DeLoughery TG, et al. The many roles of tranexamic acid: An overview of the clinical indications for TXA in medical and surgical patients. Eur J Haematol. 2020;104(2):79-87. [PubMed] | [CrossRef] | [Google Scholar]
- Satyavathi K, Naga JV, Mohammed S. Tranexamic acid: A proven antifibrinolytic agent (A review). Orient J Chem. 2009;25(4):987-92. [PubMed] | [CrossRef] | [Google Scholar]
- British Pharmacopoeia. Her majestry’s stationary office. 2010:822-823-2130-1. [PubMed] | [CrossRef] | [Google Scholar]
- The Japanese pharmacopoeia. Vol. 14. Tokyo: Ministry of Health, Labour and welfare. Society of Japanese Pharmacopoeia. 1982:1191-2. [PubMed] | [CrossRef] | [Google Scholar]
- El-Aroud KA, Abushoffa AM, Abdellatef HE. Spectrophotometric and spectrofluorimetric methods for the determination of tranexamic acid in pharmaceutical formulation. Chem Pharm Bull (Tokyo). 2007;55(3):364-7. [PubMed] | [CrossRef] | [Google Scholar]
- Ashfaq M, Aslam A, Mustafa G, Danish M, Nazar MF, Asghar MN, et al. Derivatization/Chromophore introduction of tranexamic acid and its HPLC determination in pharmaceutical formulations. J Assoc Arab Universities Basic Appl Sci. 2015;17(1):51-6. [CrossRef] | [Google Scholar]
- Chang Q, Yin OQ, Chow MS. Liquid chromatography-tandem mass spectrometry method for the determination of tranexamic acid in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;805(2):275-80. [PubMed] | [CrossRef] | [Google Scholar]
- Fiechtner BK, Nuttall GA, Johnson ME, Dong Y, Sujirattanawimol N, Oliver WC, et al. Plasma tranexamic acid concentrations during cardiopulmonary bypass. Anesth Analg. 2001;92(5):1131-6. [PubMed] | [CrossRef] | [Google Scholar]
- Vessman J, Strömberg S. Determination of tranexamic acid in biological material by electron capture gas chromatography after direct derivatization in an aqueous medium. Anal Chem. 1977;49(3):369-73. [PubMed] | [CrossRef] | [Google Scholar]
- Grassin Delyle SG, Abe E, Batisse A, Tremey B, Fischler M, Devillier P, et al. A validated assay for the quantitative analysis of tranexamic acid in human serum by liquid chromatography coupled with electrospray ionization mass spectrometry. Clin Chim Acta. 2010;411(5-6):438-43. [PubMed] | [CrossRef] | [Google Scholar]
- Abou-Diwan C, Sniecinski RM, Szlam F, Ritchie JC, Rhea JM, Tanaka KA, et al. Plasma and cerebral spinal fluid tranexamic acid quantitation in cardiopulmonary bypass patients. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(7-8):553-6. [PubMed] | [CrossRef] | [Google Scholar]
- Barreiros L, Amoreira JL, Machado S, Fernandes SR, Silva EMP, Sá P, et al. Determination of tranexamic acid in human plasma by UHPLC coupled with tandem mass spectrometry targeting sub-microgram per milliliter levels. Microchemical Journal. 2019;144:144-50. [CrossRef] | [Google Scholar]
- Crouthamel WG, Dorsch B. Specific high-performance liquid chromatographic assay for nitroglycerin in dosage forms. J Pharm Sci. 1979;68(2):237-8. [PubMed] | [CrossRef] | [Google Scholar]
- Gorynski K, Bojko B, Kluger M, Jerath A, Wąsowicz M, Pawliszyn J, et al. Development of SPME method for concomitant sample preparation of rocuronium bromide and tranexamic acid in plasma. J Pharm Biomed Anal. 2014;92:183-92. [PubMed] | [CrossRef] | [Google Scholar]
- Delavenne X, Montbel A, Hodin S, Zufferey P, Basset T. Quantification of total and unbound tranexamic acid in human plasma by ultrafiltration liquid chromatography/ tandem mass spectrometry: Application to pharmacokinetic analysis. J Pharm Biomed Anal. 2014;91:32-6. [PubMed] | [CrossRef] | [Google Scholar]
