ABSTRACT
The Quality by Design (QbD) concept has been appreciated and expected by the regulatory agencies, especially the “United States Food and Drug Administration” (USFDA), the “European Medicines Agency” (EMA), and other agencies that have adopted the “International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use” (ICH) around the globe. This paper describes the application of QbD principles in the pharmaceutical development of Oral Solid Dosage forms (OSDs). It encourages the implementation of risk-based approaches when designing pharmaceutical products. It provides a thorough understanding of formulation variables, Critical Material Attributes (CMAs), process variables, and Critical Process Parameters (CPPs) based on industrial experience that can be considered for risk assessment during the development of OSDs. It provides handy guidance to academics, research scholars, and industry scientists for implementing QbD in developing the OSDs.
INTRODUCTION
The main goal of pharmaceutical development and regulations is to ensure that each approved drug product keeps the same quality and performance characteristics throughout its commercial life as the lots that were used to prove the product’s safety and effectiveness during the approval process for marketing authorization. In the past, this assurance relied only on product specifications proposed without the support of thorough product and process understanding.1 Consequently, it is difficult to adequately control the routine product manufacturing process, leading to the failure of established quality control criteria and, equally, ineffectiveness encountered in in vivo product performance due to the lack of defining the critical quality attributes during product development. The pharmaceutical sector has evolved with the implementation of quality guidelines like ICH Q8 “Pharmaceutical Development”, ICH Q9 “Quality Risk Management”, ICH Q10 “Pharmaceutical Quality System” and ICH Q12 “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management”. It has been understood that the quality of pharmaceuticals should be designed during development and built during the manufacturing process. The majority of quality issues are related to the design of pharmaceutical products.1 No matter how many tests or studies are performed to ensure its quality, a poorly designed pharmaceutical product will exhibit low safety and efficacy. As a result, QbD starts with the understanding that increased testing of pharmaceutical items would not enhance quality. Ultimately, the product must be of high quality.2 The new paradigm focuses on the application of modern science and the use of quality risk management tools throughout the product lifecycle.3
Regulatory agencies are looking for pharmaceutical industries to create a standardized quality system that can be used throughout the product’s lifespan, with an emphasis on an integrated approach to quality risk management and science. It is expected to implement the QbD principle in pharmaceutical development. QbD is a systematic approach in pharmaceutical development that starts with predetermined objectives and highlights product and process understanding based on sound science and quality risk management.4
The QbD approach starts by identifying the Quality Target Product Profile (QTPP). The QTPP is an important element that forms the basis of the design of the product. Critical Quality Attributes (CQAs) are the properties that should be within a certain range to ensure the desired product quality. The risk assessment identifies CMAs and CPPs that have a potential impact on the CQAs of the product. The design space is a multidimensional combination and interaction of input factors and process parameters that have been shown to provide quality assurance.4 The control strategy is intended to ensure that a product with the desired quality is consistently produced.
This article is intended to discuss in-depth the pharmaceutical development of OSDs using QbD principles. OSDs like Immediate-Release (IR) and Extended-Release (ER) tablets and capsules, dispersible tablets, chewable tablets, etc. are considered in the discussion. The USFDA has published examples of pharmaceutical development studies that industries may refer to implementing QbD in their development process.5,6 There are also other guidances published recently for OSDs that have to be considered during development using QbD. The guidelines are related to the tablet scoring,7 size and shape,8 elemental impurity,9 nitrosamine,10 chewable tablets,11 drug products administered using an enteral feeding tube,12 etc.
PHARMACEUTICAL DEVELOPMENT USING QBD
The QbD elements are outlined in Figure 1 followed by a discussion of each element and their implementation in the pharmaceutical development of OSDs.
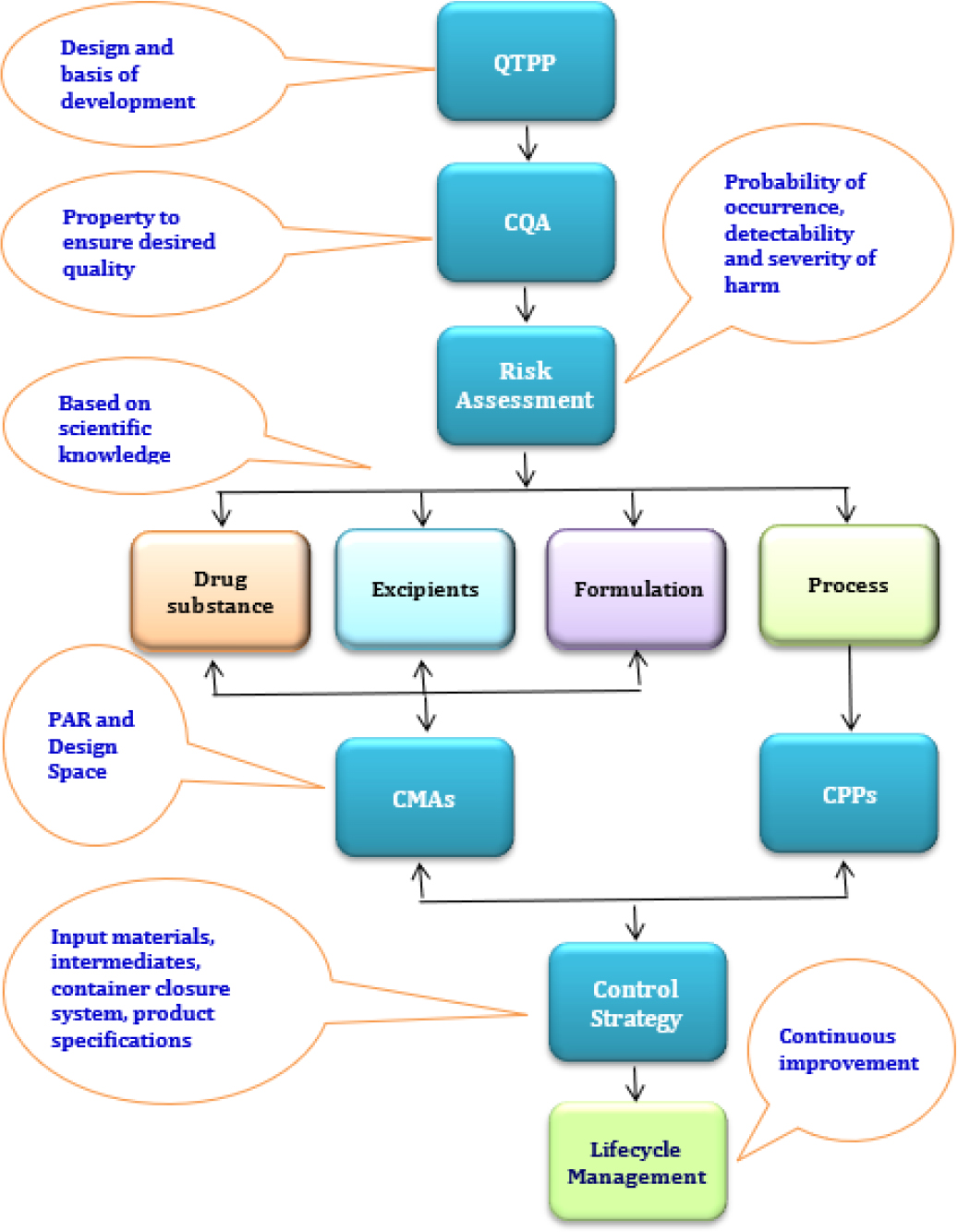
Figure 1:
QbD elements.
Quality Target Product Profile
The QTPP defines the product’s design requirements and should be used to generate CQAs, CMAs, CPPs, and a control strategy. It is concerned with quality, safety, and efficacy, taking into account factors such as route of administration, dosage forms, bioavailability, strength, and stability.4 The target should be defined before initiating the development based on the properties of the active substance, the characterization of the reference product, and the intended labeling information and population. Table 1 represents elements to consider while designing the QTPP. After setting QTPP, there is a need to identify CQAs, as discussed below.
| Elements | Target | Rationale | |
|---|---|---|---|
| Dosage form | Immediate – release tablets or capsules Delayed – release tablets Extended – release tablets or capsules Dispersible Tablets Chewable tablets | Dosage form should be same as a requirement of Pharmaceutical Equivalence. | |
| Administration | Oral route | Route of administration should be same as a requirement of Pharmaceutical Equivalence. | |
| Alternative methods of administration | Administration via enteral tube | If labeling of reference product indicates drug product’s suitability for administration via enteral tube. | |
| Dose strengths | Strength as per reference product label. | Strength should be same as a requirement of Pharmaceutical Equivalence. | |
| Pharmacokinetics | Bioequivalent to the reference product | Bioequivalence requirement. | |
| Stability | Mention desired or expected shelf life and storage condition for your proposed drug product. | Equivalent to or better than reference product shelf-life. Preferably, can be stored at room temperature. | |
| Drug product quality attributes | Physical attributes | Must meet the applicable quality standards for solid oral dosage forms. | |
| Identification | |||
| Assay | |||
| Uniformity of dosage units | |||
| Dissolution | |||
| Organic impurity | |||
| Elemental impurity | |||
| Nitrosamine impurity | |||
| Water content | |||
| Residual solvents | |||
| Container closure system | Mention desired or intended packaging system (bottle, blister, strip packaging etc.) | Drug product stability shall be ensured in proposed packaging system. | |
Critical Quality Attributes
CQA is any characteristic (physical, chemical, biological, or microbiological), that should be within a certain range. While identifying CQAs and distinguishing them from the other quality attributes, consider the severity of harm (safety and efficacy) to the patients as described in the QTPP and the basis on which it has been developed (e.g., prior knowledge, scientific principles, and experimentation).13
Drug product quality should be achieved through a quality management system and appropriate formulation design and process development. There is a need to investigate only those CQAs that are likely to be impacted by the formulation or process variables. Table 2 indicates CQAs to consider in OSDs.
| Quality attributes of the drug product | Target | Is this critical? | Justification of criticality | |
|---|---|---|---|---|
| Physical attributes | Appearance and odor | Palatable to the patients. No visual defects in the tablets or capsules. No unpleasant odor. | No | Organoleptic properties (shape and color) are not directly linked to the safety and efficacy hence, not critical. The target is set to ensure palatability otherwise it can trigger the market complaints. |
| Size | Similar or smaller than the reference product. | Yes | Tablet size correlates to swallowability; therefore, it is critical. For comparable ease of swallowing as well as patient acceptance and compliance with treatment regimens, the target for tablet size and volume is to set similar to or smaller than the reference product. USFDA guidance “Size, Shape, and Other Physical Attributes of Generic Tablets and Capsules” limits the size difference between the reference and generic drug products. | |
| Score Configuration | Similar to the reference product. | Yes | If reference product is scored and product label recommends then, generic product will be scored. | |
| Friability (whole and split tablets) | NMT 1% w/w | No | A target of NMT 1.0% can be set in-line with Pharmacopoeias. It will not impact patient safety or efficacy. | |
| Identification | +ve for active | Yes | Though identification is crucial for safety and efficacy, the quality management system can effectively regulate and monitor this CQA at the time of drug product release. Identity is unaffected by formulation or process factors. | |
| Assay (whole and split tablets) | Target to label claim | Yes | Safety and effectiveness is impacted by the assay variability. The assay of the drug product can be affected by the formulation and process factors. | |
| Uniformity of dosage units (whole and split tablets) | Conforms to Pharmacopoeial general chapter <Uniformity of dosage units> | Yes | Safety and effectiveness is impacted by the content variability. The content uniformity can be affected by the formulation and process factors. | |
| Dissolution | Whole Tablets | To comply the drug release specifications which are derived based on the drug release of the batches used in clinical or bioequivalence study using a discriminatory dissolution method. | Yes | In vitro dissolution is linked with the in vivo absorption and performance. Both formulation and process variables can affect the drug release. |
| Split Tablets | Similar drug release to that of whole tablets. | |||
| Residual solvents | Conforms to Pharmacopoeial general chapter limits and ICH Q3C. | Yes | Residual solvents can impact patient safety. Depending on the solvents used in the manufacturing, process variables can impact this CQA. Product CQA is unlikely to be affected by formulation or process variables if no solvents used in the manufacturing. | |
| Impurity | To comply product specific monographs or ICH Q3B. | Yes | Failure to comply the impurity requirement might jeopardize safety. Degradants impurity profile can be affected by the both formulation and process variables. | |
| Water content | Set based on the trend data or theoretical carryover from the all raw materials used in the drug product. | Yes | Water content, in general, can impact hydrolytic degradants and the microbial growth, and therefore be a possible CQA. | |
| Alcohol-induced dose dumping (ER drug products) | No dose dumping or comparable to the reference product. | Yes | The target is to ensure no burst release is induced by the alcoholic medium, which can produce additional risks to the patient. | |
| Microbial Limits | Conforms to Pharmacopoeial general chapter limits. | Yes | Microbe impacts patient safety. If raw materials comply with microbial requirements, the formulation and process variables are unlikely to impact this CQA. However, depending on the nature of excipients and manufacturing process used it can impact this CQA. | |
| Elemental Impurity | Conforms to Pharmacopoeial general chapter limits or ICH Q3D. | Yes | Elemental impurities can impact safety. If input materials, packaging materials, manufacturing equipment, and utilities are controlled to comply with elemental impurity, this CQA is unlikely to be affected by formulation or process variables. | |
| Nitrosamine Impurity | Conforms to ICH M7 guidance. | Yes | Nitrosamine impurity can impact safety. Presence of amine groups in the drug molecule or the excipients can react with nitrates/nitrites under acidic conditions leads to formation of nitrosamine impurities. If actives and excipients do not have the potential to generate nitrosamine impurities, this CQA is unlikely to be affected by formulation or process variables. | |
RISK ASSESSMENT
Risk includes the probability of occurrence, the detectability, and the degree of harm. Therefore, using a risk management system, the level of risk can be changed. The following are some principles to keep in mind while adopting quality risk management.5,6
The assessment of the risk to quality should be based on scientific evidence and ultimately connected to patient safety.
The relative risk of each element can be ranked as high, medium, or low. Further investigation is required for those elements likely to have a high impact on the drug product CQAs. No further investigation is required for the elements having a low impact. A similar relative ranking system can be used throughout product development.
During pharmaceutical development, risk assessment can be carried out in three stages: risk assessment of drug substance attributes, formulation components, and the drug product manufacturing process, as discussed below.
RISK ASSESSMENT OF DRUG SUBSTANCE ATTRIBUTES
Risk assessment of drug substance attributes on drug product CQAs can be performed based on the physical, chemical and, biological properties of the drug molecule.4 Drug substance attributes and drug product CQAs enlisted in Table 3 shall be considered during risk assessment. The rationale for assigning the risk is explained further based on the literature available and the knowledge gained from industrial experience.
| Drug product CQA | Drug Substance Attributes |
|---|---|
| Physical attributes (Tablets or capsules) Tablet splitability (scored tablets) Assay Content Uniformity Dissolution (whole tablets; split tablets in case of scored tablets) Degradants Elemental Impurity Nitrosamine Impurity Microbial Count | Particle size Solid state form Solubility Flow properties Organic impurities Elemental impurities Residual solvents Microbial Count Hygroscopicity |
Particle size
The risk of particle size impacting physical attributes can be assigned based on the contribution of the drug substance in the final formulation and the manufacturing process chosen (direct compression or granulation). Drug substances having a significant contribution to the final formulation can play a major role in the physical attributes through their compressibility and flow properties.14 With a low concentration of drug substance in the formulation, particle size distribution can have a direct impact on drug distribution and, ultimately, on the assay and content uniformity.15 The risk can be high or moderate, depending on the manufacturing process adopted. Consequently, risk can be assigned. The effect of particle size on dissolution depends essentially on the solubility class of the drug substance as per the Biopharmaceutical Classification System (BCS). The risk of particle size impact dissolution can be higher with poorly soluble drugs.16 There is no role for particle size if drugs are added in solution form, neither in assay and content uniformity nor in dissolution. The risk of particle size influencing the dissolution of whole tablets is directly related to the dissolution of split tablets. If a drug substance is stable, particle size will not affect degradants or nitrosamine impurities. There is no relationship between particle size and elemental impurities in the drug product.
Solid state form
Drug substances that exhibit polymorphism can impact drug product CQAs. The compressibility and subsequent physical properties of tablets can be influenced by solid-state form.17,18 Crystal nature can affect flow properties and, later, content uniformity. Different polymorphic forms can have different solubility, dissolution and rate of degradation; risk can be performed based on the polymorphic stability by the drug manufacturer. While performing the risk assessment, USFDA guidance (ANDAs: pharmaceutical solid polymorphism) can be referred.19
Drug solubility
The probability of drug solubility having an impact on drug product CQAs is low, except for dissolution. Depending on the solubility class and pH-dependent solubility behavior of the drug molecule, a risk assessment can be performed.
Flow property
The flow property of the drug substance matters when the manufacturing process is direct compression to the tablets or direct filling into capsules, and it has a significant contribution to the final blend. It can impact physical attributes like tablet or capsule weight variation, assay, and content uniformity.15 The flow property of the drug is diminished using the granulation process. The risk of flow property impacting physical attributes, assay, and content uniformity can be assessed depending on the manufacturing process chosen. It plays no role in the generation of degradants or other impurities (elemental and nitrosamine).
Organic impurities
The physical attributes of the product are not linked to organic impurities. Impurities usually do not impact a drug product’s assay, content uniformity, and dissolution if their presence and level are controlled in the drug substance specification. The risk to impact degradants in the drug product can be determined if there is evidence of incompatibility or conversion of drug process impurities to the degradants. If the route of drug synthesis or any impurities put any risk of nitrosamine impurity in the drug product, the risk can be assigned and investigated accordingly. Processes using nitrites in the presence of acids, secondary, tertiary, or quaternary amines can create the risk of generating nitrosamine impurities.10
Elemental impurities
The physical attributes of the product are not linked to the elemental impurities. The risk assessment must be carried out to evaluate elemental impurities carryover from the drug substance to ensure that the final drug product does not cross Permitted Daily Exposure (PDE) as per United States Pharmacopoeia (USP) general chapter20 <232> and ICH Q3D guidance.9 The risk of elemental impurities can be higher in the case of inorganic drug molecules (e.g. potassium chloride, sodium chloride), depending on the risk, the control strategy can be in-placed. The risk of the impact of elemental impurities to impact assay, content uniformity and dissolution is low. Depending on the evidence of element-catalyzed degradation in drug products, the risk of impacting finished product degradants can be assigned.21,22
Residual solvents
If the level of solvents is controlled in the drug substance in-line with option 1 of USP general chapter <467> or ICH Q3C guidance and the total daily intake of the drug product is less than 10 g, then the risk is low. If residual solvents exceed the option 1 limit, carryover in drug products must be calculated based on the total daily drug product intake.23,24 If any solvent is present at a significant level, risk can be evaluated considering its role in the degradation pathways of the drug product.
Microbial count
Depending on the water activity and route of synthesis of the drug substance, it may support microbial growth.25 As per pharmacopoeia and regulatory expectations, there is a need to control total aerobic microbial count, total combined yeast and mould count, and specified micro-organism – Escheria coli in the raw materials going to use in oral solid dosage forms.26,27 The risk assessment shall be done and control has to be given through raw material specifications.
Hygroscopicity
It is an important element to consider in risk assessment. Hygroscopic drug substances can impact process feasibility, physical attributes, degradants, and microbial count.28 Depending on the manufacturing process, environmental conditions for the operations shall be selected.
After the risk assessment of drug substances, the selection of excipients and their grades, followed by drug-excipient compatibility, is an important step for understanding the role of inactive ingredients in product quality. The selection of raw materials and their grades for the compatibility study should be based on prior knowledge about the drug, its impurities, and degradation pathway, as well as the further manufacturing processing conditions to be used for the drug product. A scientifically sound approach should be used in constructing compatibility studies.
RISK ASSESSMENT OF FORMULATION COMPONENTS
Ideally, product development is divided into two stages: formulation development and process development. At the time of the risk assessment of formulation components, an optimized manufacturing process may not be established, and risks can be analyzed and evaluated using a preliminary manufacturing process. It shall begin with the overall risk assessment of the drug product components to determine which components are likely to have a high risk of impacting the drug product’s CQAs. Depending on the drug product and dosage form, formulation components can be considered in the risk assessment.4 The formulation components for guidance enlisted in Table 4 should be considered during risk assessment. The rationale for assigning the risk is explained further.
| Drug product CQA | Immediate release dosage forms | Extended- release dosage forms |
|---|---|---|
| Physical attributes (Tablets or capsules) Assay Uniformity of dosage units Dissolution (whole tablets; half tablets in case of scored tablets) Degradants Elemental Impurity Nitrosamine Impurity Microbial Count Alcohol Dose Dumping (Modified release dosage) Dispersion Time (Dispersible Tablets) Hardness (Chewable Tablets) Disintegration Time (Conventional tablets, Chewable Tablets) | Drug Particle size Diluent/Filler type and ratio Binder type/level Disintegrant type/ level Surfactant/wetting agent type/level Glidant level Lubricant type/level Stabilizer/ Preservative type/level | Matrix Technology: Drug Particle size Diluent/Filler type and ratio Lubricant type/level Glidant level Release Controlling Polymer type/level Release Controlling Polymer Polymer lot to lot variability Reservoir Technology: Drug Particle size Diluent/Filler type and ratio Lubricant type/level Release Controlling Polymer type/level Release Controlling Polymer lot to lot variability Pore former type/level Plasticizer type/level Solvent type Core pellet size Cushioning agent level |
Particle size
As discussed under the section risk assessment of drug substance, depending on the contribution of drug substance in the final formulation, selected manufacturing process and solubility class risk of particle size to impact drug product CQA can be assigned.
Diluent or filler type and ratio
The diluent can be used alone or in combination. Diluent type and level can significantly impact physical attributes like size, weight, and hardness, including tablet splitability.29,30 Depending on the dose, particle size of the drug substance, and the manufacturing process, risk assessment can be performed and diluent grades can be selected. The type of dosage form, like dispersible and chewable tablets, shall be given special attention considering palatability due to the use of a particular diluent, and the same shall be considered during risk assessment. Diluent’s particle size and flow properties are important to achieve assay and content uniformity. Lactose incompatibility with drugs having secondary amines,31 diluents having high moisture content or peroxides should be considered during risk assessment to impact degradants depending on the drug molecules.
Binder type and level
Binder type and level can have a direct impact on compressibility, granule properties, tablet hardness, splitability, disintegration, dispersion time, and dissolution.32 Depending on the solubility class of the drug, the risk of binder to impact dissolution can be assigned. The risk of binders to impact degradants can be assigned based on the drug-excipients compatibility study or any known interactions.
Disintegrant type and level
The disintegrant promotes disintegration either by water wicking (crospovidone) or swelling (croscarmelose sodium and sodium starch glycolate).33 Disintegrant types are important to mimic the disintegration behavior of the generic formulations with the reference product. Level of disintegrant can impact the disintegration time and later dissolution. However, it can affect the stability of the formulation due to its water-loving and absorption tendency (for example, use of crospovidone with diluent dicalcium phosphate dihydrate or trihydrate). Crospovidone rapidly absorbs water. At accelerated stability storage conditions, dicalcium phosphate loses water, which allows it to swell, resulting in tablet/capsule defects, loss of tablet integrity, hardness etc.34
Surfactant or wetting agent type and level
It is preferred to use a minimum effective level of surfactant or wetting agent with poorly soluble drugs. Risk assessment should be conducted depending on the drug molecule and different types of surfactants like cationic, anionic, or nonionic. Surfactants or wetting agents can impact the physical attributes e.g., docusate sodium is used as a wetting agent, which is a waxy material that impacts tablet compressibility significantly. Polysorbates are liquid surfactants that cannot be used in direct compression or direct capsule filling processes; and if used in a granulation process, the risk of affecting physical attributes must be assessed. The risk of surfactants to impact the degradants shall be assessed based on the sensitivity of the drug molecules.
Glidant level
Colloidal silicon dioxide and talc are commonly used glidants in solid oral dosage forms. Its level may or may not have a significant impact on the drug product CQAs; however, risk assessment must be performed to determine the optimum level of the glidant. Significantly low and high concentrations have a negative effect on the flow properties.35
Lubricant type and level
Lubricant can be considered the most significant element of solid oral dosage forms. Magnesium stearate is the choice of lubricant in the pharmaceutical industry due to its effectiveness. Depending on the risk of using magnesium stearate, other lubricants like stearic acid, glyceryl behenate, and sodium stearyl fumarate can be tried. Lubricant type and level can have a significant impact on tablet and capsule physical properties, dissolution, and sometimes degradants (magnesium can act as a catalyst in degradation,36 and sodium stearyl fumarate can form adducts with certain molecules.37,38 Lubricant levels can have a high risk of impacting the dissolution of poorly soluble drugs from IR dosage forms as compared to ER dosage forms.
Stabilizers type and level
In OSDs, stabilizers are added to prevent degradation due to acid, base, peroxides, hydrolysis, or oxidation-reduction reactions. Preservatives can be added to control microbial growth. The selection of stabilizer should be based on the underlying mechanism of degradation in the drug product. The risk assessment shall be performed to finalize and justify the minimum effective level of stabilizers or preservatives to ensure the drug product’s CQA (degradants/microbial count) remains within acceptance criteria during batch release and throughout the shelf life.
Release-controlling polymer type and level
Commonly, modified release formulations are designed using matrix or reservoir technology. Release-controlling polymers play important role in achieving the product CQAs. In the matrix technique, the risk of type and level of the polymer to impact physical attributes, assay, content uniformity and dissolution shall be evaluated. In the reservoir technique, the type and level of the polymer have a more direct impact on dissolution than any other drug product CQA, risk shall be assigned accordingly. Performance of delayed-release or timed-release dosage forms largely depends on the polymers having pH-dependent solubility; the risk of polymers levels to resist the drug release in an acidic environment shall be evaluated critically. In all modified release formulations, the risk of the type of polymers to impact alcohol dose dumping shall be also evaluated. Natural-originated cellulosic polymers can contain nitrites which can support the formation of nitrosamine impurities in the drug molecule having secondary to quaternary amine groups.10
Release controlling polymer variability
Lot-to-lot or vendor-to-vendor variability signifies the CMAs of the polymer. It is important to perform risk assessment considering the lot-to-lot and or vendor-to-vendor variation which may cause due to the difference in CMAs although it is within the specifications having wider limits. It is also necessary to evaluate the polymer performances vendor-to-vendor due to their different manufacturing process or control strategies. CMAs for the release-controlling polymers are polymer viscosity, particle size, hydroxy – propoxy content which can be thoroughly investigated at their extreme specifications as the risk of impact dissolution is high; if needed tighter in-house control over CMAs can be provided.39
Plasticizer type and level
Plasticizers are added to modify the film properties of the release-controlling polymer.40 The choice of plasticizer (hydrophilic nature, hydrophobic nature, type) depends on the type of polymer used.41 The risk of plasticizer to impact dissolution and alcohol dose dumping is higher, investigation shall be done accordingly.
Pore former level
Pore formers are added to hydrophobic polymers to modify and achieve the desired drug release. The level of pore formers in the drug product is critical to maintaining the dissolution; small changes in the pore former can lead to batch-to-batch variation due to its high solubility in aqueous media.
Solvent types
In solid dosage forms, solvents can be used for granulation, tablet coating, and pellet coating. The risk of solvent type to impact dissolution from the modified release dosage forms is always higher; it can be moderate for IR dosage forms. The risk of solvent to impact degradants can be evaluated based on the route of degradation; sometimes it can also lead to the polymorphic conversion of drug molecules.
Core pellet size
The selection of core pellet size is important during generic product development to mimic the drug release profile of the reference product. It also impacts the friability during operation, which can impact the assay and sometimes the content uniformity of the low-dose molecules during capsule filling. The risk of core pellet size to impact assay and content uniformity is also high where polymer-coated pellets are compressed to the tablets using cushioning agents; a significant difference in the pellet size distribution and density can lead to segregation. The risk of pellet size to impact assay, content uniformity, and dissolution can be assessed accordingly.
Cushioning agent level
Cushioning agents are added to the dispersible or micro- dispersible tablets containing multiparticulate. After dispersion, the drug release is controlled by the polymeric film of multiparticulate. Cushioning agents provide cushion to the multiparticulate during compression, preventing rupturing of the polymeric film. The risk of cushioning agents impact on physical attributes (tablet size, compressibility, and splitability), content uniformity, and dissolution shall be evaluated.42
After completion of the initial risk assessment of the formulation variables, high-risk factors should be considered for further investigation; in some cases, variables considered medium-risk can also be investigated. Experiments can be designed to study One Factor at A Time (OFAT) or use a multifactorial design. The factors shall be the formulation variables and the responses shall be the CQAs or intermediate CQAs. The formulation risk assessment identifies potential high-risk CMAs which can impact product CQAs and should be part of the control strategy. Depending on the function of input materials in the drug product, CMAs can be as follows: as shown in Table 5.
| Input Materials | Critical Material Attributes (CMAs) |
|---|---|
| Drug substance | Particle size, Solid state, Hygroscopicity, Solubility, Impurities (organic, elemental and nitrosamine), bulk density. |
| Diluents | Particle size, moisture content, bulk density, microbial count. |
| Binders | Viscosity, peroxide contents. |
| Disintegrant | Water soluble/insoluble contents. |
| Glidant | Particle size and specific surface area. |
| Surfactant | HLB values. |
| Lubricant | Particle size and specific surface area. |
| Release controlling polymer | Viscosity, Particle size, Ethoxy contents, Hydroxy-propoxy contents. |
| Core Pellets | Size distribution, friability, components. |
| Solvents | Elemental impurities, organic impurities. |
Risk Assessment of Drug Product Manufacturing Process
To identify the high-risk steps that are likely to impact the final drug product CQAs, a risk assessment of the manufacturing process should be performed while developing and optimizing the manufacturing process. Following that, identify the intermediate CQAs that are directly connected to the final drug product CQAs. The risk assessment should focus on the process variables that potentially affect the selected intermediate CQAs to identify which process factors have the highest probability of causing CQA failure. To improve the manufacturing process and decrease the chance of failure, those variables should be evaluated.4 Refer Table 6 for different manufacturing steps depending on the type of formulation which can impact intermediate CQAs and later impact final drug product CQAs. The rationale for assigning the stepwise risk is explained further.
| Manufacturing steps | Intermediate CQAs | Final Drug Product CQAs |
|---|---|---|
| Mixing: Pre-lubrication | Blend Assay, Blend Uniformity. | Assay, Content uniformity. |
| Mixing: lubrication | Physical attributes, dissolution. | Description, Dissolution. |
| Granulation | Particle size distribution, degradation during hold time, Loss on Drying. | Content uniformity, Dissolution, Splitability of scored tablets, Degradants, Microbial count, Residual solvents. |
| Milling | Particle size distribution. | Dissolution, Splitability of scored tablets, Assay, Content uniformity. |
| Roller Compaction or Slugging | Particle size distribution, Blend Uniformity. | Dissolution, Splitability of scored tablets, Assay, Content uniformity. |
| Compression | Weight variation, Hardness, Friability, Disintegration time, Content uniformity, Dissolution, Assay. | Description, Assay, Content uniformity, Dissolution, Splitability of scored tablets. |
| Film Coating | Appearance, hold time of coating solution, Disintegration time. | Description, Microbial count, Dissolution. |
| Drug layering/ coating | Assay, hold time of drug solution. | Assay, Content uniformity, Degradants, Microbial count, Residual solvents, Residual solvents. |
| Controlled Release Polymer Coating | Dissolution. | Dissolution, Residual solvents. |
| Capsule Filling | Weight variation, Content uniformity, Assay. | Assay, Content uniformity. |
Mixing-pre-lubrication
The particle size and cohesiveness of the drug substance can adversely impact its flowability, which can affect intermediate CQAs like blend uniformity and blend assay, which later results in the failure of drug product CQAs like assay and content uniformity.14,15 The risk of the blending process to impacting these CQAs is less if drug substance is added in the granulation step and the concentration of extra granular components is insignificant. The risk of the pre-lubrication/blending step impacting other attributes like physical attributes, dissolution, and degradation is low. Holding the blend can also lead to segregation and impact the blend uniformity.
Mixing-lubrication
The lubrication time can impact the compressibility and hardness of the tablet.43 The lubricant type, concentration, lubrication time, and specific surface area are the key elements in a successful compression process. Poor lubrication results in sticking/picking/ ejection problems, and over-lubrication results in capping/ lamination issues during compression. Poor lubrication can also impact capsule filling due to sticking to the tamping pins. Depending on the solubility class of the drug, lubrication time can impact dissolution.44 The probability of drugs with high aqueous solubility being impacted by lubrication is lower; it can be high in the case of poorly soluble drugs.
Granulation
Granulation is performed to improve flow, compressibility, and uniformity.45 Depending on the type of diluent and drug concentration, poor or over-granulation can affect intermediate CQAs like particle size distribution, which later impact compression parameters like weight variation, hardness, tablet splitability, content uniformity, and dissolution. Loss on drying after the granulation stage is critical for controlling blend compressibility, residual solvents (if organic solvents are used for granulation), microbial load, and degradation of sensitive molecules (by hydrolysis).46
Milling
Milling is performed to achieve uniform granules and size distribution. Particle size distribution can be considered an intermediate CQA as it affects blend uniformity, weight variation, hardness, tablet splitability, content uniformity, and dissolution. The probability of a milling step impacting degradants is lower unless the drug is heat sensitive or has a low melting point affected by the heat generated during milling.
Roller compaction
Like any granulation, roller compaction or slugging is performed to improve flow, compressibility, and uniformity, and, additionally, prevent degradation due to solvents. The particle size distribution is a critical in-process factor to control during the dry granulation process as it later plays a role in achieving finished product CQAs like physical attributes, tablet splitability, content uniformity, and dissolution.5
Compression
The shape, size, and score configuration of the tablets are defined by the tooling design selected during development. Compression force can impact the tablet hardness, splitability of the scored tablets, friability, disintegration time, and dissolution. Press speed and feeder speed can impact tablet weight variation and content uniformity. Sometimes, tablet weight variability can lead to Out-of-Specification (OOS) assay results. The compression is unlikely to impact degradation.5,6
Film coating
Coating defects can impact the elegancy of the finished dosage form. Assay and content uniformity are mainly determined up to the compression step and are unaffected by the film coating process variables. The risk of the film coating process impacting tablet assay and content uniformity is low. The film coating composition can sometimes moderately impact the disintegration time of the coated tablets and later dissolution. The hold time of aqueous film coating dispersion can impact the microbial count in the drug product. Depending on the type of solvent used in the coating process, and the sensitivity of the drug to the particular solvent, it can impact degradants.
Drug layering/coating
Drug layering over multiparticulate is done using a wurstor fluid bed processor, and over tablets is done using conventional coating pans. In both cases, the risk of this step to impact the assay is high.6 In the case of multiparticulate risk to impact, content uniformity can be lower than in the tablet pan coating. The hold time of a drug coating solution can impact the degradants and microbial count.
Controlled-release polymer coating
Controlled-release polymer coating, either performed on multiparticulate or tablets, directly impacts dissolution and alcohol-induced dose dumping. The risk of affecting other products’ CQAs is less.6
Capsule filling
The risk of the capsule filling step to impact weight variation and later content uniformity is high. Sometimes improper setting can lead to capsule defects, but the risk of impacting other product CQAs is lower.
The drug product manufacturing process risk assessment identifies potentially high-risk attributes and/or process parameters. Depending on the risk assigned, design and conduct experiments to determine which process parameter is critical. Depending on the manufacturing process and the type of drug product, CPPs can be established (Table 7).
| Manufacturing steps | Equipment Variables | Critical Process Parameters (CPPs) |
|---|---|---|
| Powder Mixing | Type of blender, mixing principle. | Blender rotational speeds, number of revolutions, material load, hold time. |
| High Shear Granulation | Type of granulator. | Material load, granulation time, impeller speed, fluid uptake, end point impeller current. |
| Top Spray Granulation | Nozzle diameter. | Material load, fluidization air, spray rate, atomization air. |
| Drying | Type of dryer. | Material load, inlet temperature, exhaust temperature, loss on drying, fluidization air. |
| Milling | Type of Mill, milling principle. | Mill speed, screen size. |
| Roller Compaction | – | Roll speed, Roll gap, Roll force and Feeder speed. |
| Compression | Tooling type, single rotary/double rotary, gravity feeder/force feeder. | Tablet press speed, feeder speed, compression force, pre-compression fore, dwell time, ejection force. |
| Pan Film Coating | Pan design, pan size, nozzle diameter, type and number of spray guns. | Pan load, inlet temperature, exhaust temperature, pan speed, spray rate, atomization air, and coating solution viscosity, hold time of coating solution, coating weight gain. |
| Drug layering/ coating using wurstor Process | Equipment type, wurstor insert diameter, partition column diameter, air distribution plate, nozzle diameter, number of spray guns, filter type and aperture size. | Equipment load, partition height, inlet temperature, exhaust/ outlet temperature, spray rate, atomization air, air volume, coating solution viscosity, hold time of coating solution, drying time, coating weight gain. |
| Controlled Release Polymer Coating | Equipment type, wurstor insert diameter, partition column diameter, air distribution plate, nozzle diameter, number of spray guns, filter type and aperture size. | Equipment load, partition height, inlet temperature, exhaust/ outlet temperature, spray rate, atomization air, air volume, coating solution viscosity, hold time of coating solution, drying time/curing time, coating weight gain. |
| Capsule Filling | Loading plate size. | Tamping speed, feeder speed. |
Design space
The study of any individual unit operation or parameter while keeping other parameters constant, will give a Proven Acceptable Range (PAR). By changing more than one factor at a time, multidimensional combinations and interactions of input variables and process parameters can be evaluated. If it demonstrates its ability to provide assurance of quality, it generates design space.4 Accordingly, develop a control strategy by controlling CMAs, fixing the processing equipment, and defining acceptable ranges for the CPPs.
Control strategy
A control strategy is a planned set of controls (Figure 2) derived from formulation and process understanding to ensure that the desired quality is consistently produced.4 The elements of the control strategy can include the critical attributes related to the input materials (drug substances and excipients), intermediates (in-process semi-finished goods), container closure systems, product specifications, and testing or monitoring frequencies that contribute to the final product quality. The control strategy can be further reviewed and revised based on additional experience gained or as part of continuous improvement during the commercial lifecycle of the product. Working within the design space is not considered a change; moving out of the design space is considered a change and requires regulatory approval as per post-approval change procedures.
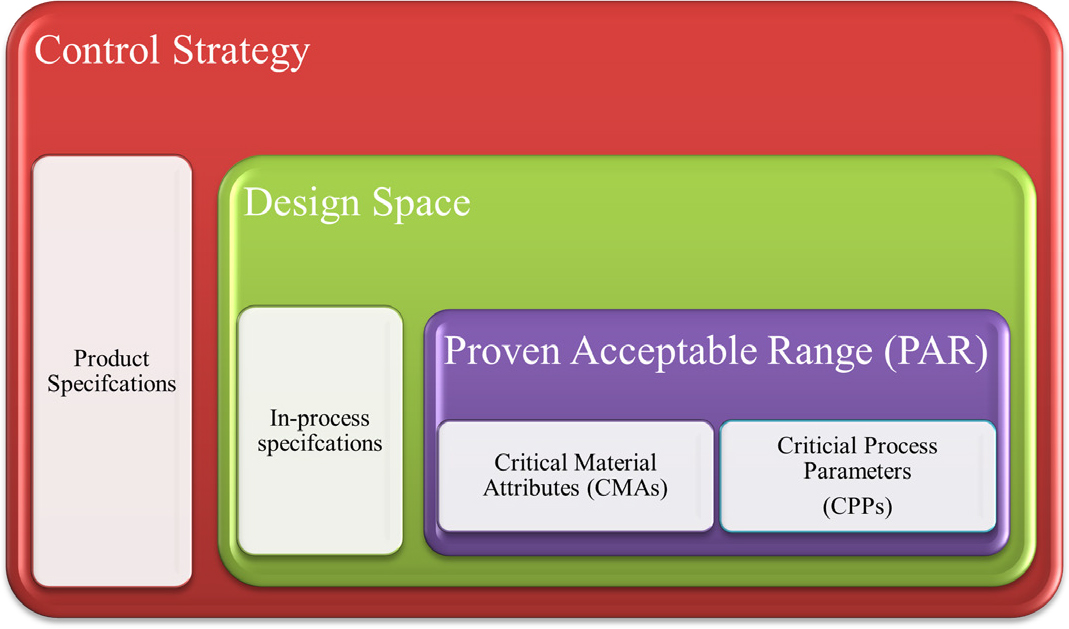
Figure 2:
Outline of control strategy.
Product lifecycle management
As a part of continuous improvement, it is expected of pharmaceutical companies to evaluate innovative methods to improve product quality throughout the commercial life of the product. Product performance can be monitored and verified for its robustness or anticipated quality through trend data analysis from product annual reports. Recently, ICH also published guidance Q12- “Technical and regulatory considerations for pharmaceutical product lifecycle management”; which provides a context for simplifying the management of post-approval changes in a more efficient manner using technical and risk-based approaches. This guideline also demonstrates how increased product and process understanding can be helpful in managing post-approval changes.47 Risk-based categorization of post-approval changes, including the identification of established conditions, will enable to making decisions regarding whether the proposed changes require notification to the regulatory agency or not, or require prior approval.
CONCLUSION
Based on existing guidance and references, QbD is an essential part of pharmaceutical quality. It is intended to provide a better understanding of the product, process, risk, and mitigation. The QbD approach leads to the development of robust products, more efficient technology transfers, and minimizes batch failures. The QbD approach allows for even more regulatory flexibility in the future. Ease of dealing with Chemistry, Manufacturing, and Controls (CMC) within a pre-existing design space, reduction in post-approval changes, time savings, and cost-effective commercialization.
References
- Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771-83. [PubMed] | [CrossRef] | [Google Scholar]
- Barshikar R. Quality by Design (QbD) and its implementation in pharma industry. Express pharma. 2013 [CrossRef] | [Google Scholar]
- . Pharm Qual Syst. 2008;Q10 [CrossRef] | [Google Scholar]
- . Pharm Dev. 2009;Q8(R2) [CrossRef] | [Google Scholar]
- Pharmaceutical development report example QbD for IR generic drugs. 2012
- Pharmaceutical development report example QbD for MR generic drugs. 2011
- USFDA guidance for industry. Tablet scoring: nomenclature, labeling, and data for evaluation. 2013
- USFDA guidance for industry. Size, shape, and other physical attributes of generic tablets and capsules. 2022
- ICH harmonised tripartite guideline. Guideline for elemental impurities. 2019;Q3D(R1)
- USFDA guidance for industry. Control of nitrosamine impurities in human drugs. 2021
- USFDA guidance for industry. Quality attributes consideration for chewable tablets. 2018
- USFDA guidance for industry. Oral drug products administered via enteral feeding tube: testing and labeling recommendations. 2021
- USFDA guidance for industry. 2012;Q8(Q9):Q10 [CrossRef] | [Google Scholar]
- Kolodziejczyk K. Effect of API on powder flowability, direct compression and properties of orally disintegrating tablets. Indian J Pharm Sci. 2019;81(3):489-95. [CrossRef] | [Google Scholar]
- Jakubowska E, Ciepluch N. Blend segregation in tablets manufacturing and its effect on drug content uniformity – a review. Pharmaceutics. 2021;13(11):1909 [PubMed] | [CrossRef] | [Google Scholar]
- Sandri G, Bonferoni MC, Ferrari F, Rossi S, Caramella CM. The role of particle size in drug release and absorption. Part Technol S. 2014;19:323-41. [CrossRef] | [Google Scholar]
- Rojas J. Effect of polymorphism on the particle and compaction properties of microcrystalline cellulose. Cellulose – medical, pharmaceutical and electronic applications. Intech open. 2013 [CrossRef] | [Google Scholar]
- Hadzović E, Betz G, Hadzidedić S, El-Arini SK, Leuenberger H. Roller compaction of different pseudo polymorphic forms of theophylline: effect on compressibility and tablet properties. Int J Pharm. 2010;396(1-2):53-62. [PubMed] | [CrossRef] | [Google Scholar]
- . Chemistry, manufacturing, and controls information. 2007 ANDAs: pharmaceutical solid polymorphism.
- . General chapter <232> Elemental impurities – limits. 2023;USP43-NF38:6641 [PubMed] | [CrossRef] | [Google Scholar]
- Sheth AC, Patel PU. Review of elemental impurities in pharmaceuticals arena. Int J Pharm Qual. 2020;11(2):214-8. [CrossRef] | [Google Scholar]
- Gabrič A, Hodnik Ž, Pajk S. Oxidation of drugs during drug product development: problems and solutions. Pharmaceutics. 2022;14(2):325 [PubMed] | [CrossRef] | [Google Scholar]
- . General chapter <467> Residual solvents. 2023;USP43-NF38:6712 [PubMed] | [CrossRef] | [Google Scholar]
- ICH harmonised tripartite guideline. Guideline for elemental impurities. 2019;Q3C(R8)
- Allen LV. Quality control: water activity considerations for beyond-use dates. Int J Pharm Compd.. 2018;22(4):288-93. [PubMed] | [Google Scholar]
- . General chapter <1111> Microbiological examination of non-sterile products: acceptance criteria for pharmaceutical preparations and substances for pharmaceutical use. 2023;USP43-NF38:7819 [PubMed] | [Google Scholar]
- . General chapter <1112> Application of water activity determination to non-sterile pharmaceutical products. 2023;USP43-NF38:7820 [PubMed] | [Google Scholar]
- Arigo A, Jawahar N, Nikhitha K, Jubie S. Effect of hygroscopicity on pharmaceutical ingredients, methods to determine and overcome: an overview. J Pharm Sci Res. 2019;11(1):6-10. [PubMed] | [Google Scholar]
- Uhumwangho MU, Okor RS. A comparative study of the dissolution characteristics of capsule and tablet dosage forms of melt granulations of paracetamol – diluent effects. Acta Pol Pharm. 2007;64(1):73-9. [PubMed] | [Google Scholar]
- Roslan NSF, Bin LK, Bostanudin MF, Razak FSA. Comparison of various fillers on the physical properties of compounded tablets. J Pharm Negat. 2022;13(7):125-32. [PubMed] | [Google Scholar]
- Bharate SS, Bharate SB, Bajaj AN. Interactions and incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: a comprehensive review. J Excipients Food Chem. 2010;1(3):1-26. [PubMed] | [Google Scholar]
- El-Setouhy DA, Basalious EB, Abdelmalak NS. Effect of different meltable binders on the disintegration and dissolution behavior of zolmitriptan oromucosal fast melt tablets. J Pharm Nutr Sci. 2017;7(1):13-23. [CrossRef] | [Google Scholar]
- Desai PM, Liew CV, Heng PWS. Review of disintegrants and the disintegration phenomena. J Pharm Sci.. 2016;105(9):2545-55. [PubMed] | [CrossRef] | [Google Scholar]
- Colorcon technical data; dibasic calcium phosphate replacement with starch. Vol. 2007; 1500® in a direct compression formula.
- Advankar A, Maheshwari R, Tambe V, Todke P, Raval N, Kapoor D, et al. Specialized tablets: ancient history to modern development. Drug delivery systems. Adv Pharm Prod Dev Res. 2019:615-64. [PubMed] | [CrossRef] | [Google Scholar]
- D’cruz D, Ajith A, Subbaraj T, Kamalasanan K. Magnesium stearate is an incompatible excipient for aspirin in wet granulation producing non-linear degradation. J Pharm Sci Res. 2018;10(2):240-2. [PubMed] | [CrossRef] | [Google Scholar]
- Ludvigsson JW, Wikström H, Andersson T, Norrby PO. Degradation caused by incompatibility between sodium stearyl fumarate (PRUV) and AZD7986 in the drug product. J Pharm Biomed Anal. 2018;158:82-7. [PubMed] | [CrossRef] | [Google Scholar]
- . Sitagliptin tablets. 2023;USP43-NF38:4051 [PubMed] | [CrossRef] | [Google Scholar]
- Deng H, Prusak B, Martin L, Vass S, Missaghi FTP. American Association of Plastic Surgeons. 2012 Critical material attributes consideration for extended release propranolol HCl hydrophilic matrix tablets.
- Porter S, Sackett G, Liu L. Development, optimization, and scale-up of process parameters: pan coating. Developing solid oral dosage forms. Pharmaceutical Theory and Practice. 2017:953-96. [PubMed] | [CrossRef] | [Google Scholar]
- Somwanshi SB, Dolas RT, Wagh VD, Kotade KB. Pharmaceutically used plasticizers: a review. European j biomed Pharm. 2016;3(2):277-85. [PubMed] | [CrossRef] | [Google Scholar]
- USFDA product label, Klor-Con M (potassium chloride extended-release tablets, USP).
- Kikuta J, Kitamori N. Effect of mixing time on the lubricating properties of magnesium stearate and the final characteristics of the compressed tablets. Drug Dev Ind Pharm. 1994;20(3):343-55. [CrossRef] | [Google Scholar]
- Abe H, Otsuka M. Effects of lubricant-mixing time on prolongation of dissolution time and its prediction by measuring near infrared spectra from tablets. Drug Dev Ind Pharm. 2012;38(4):412-9. [PubMed] | [CrossRef] | [Google Scholar]
- Shanmugam S. Granulation techniques and technologies: recent progresses. BioImpacts. 2015;5(1):55-63. [PubMed] | [CrossRef] | [Google Scholar]
- Thapa P, Lee AR, Choi DH, Jeong SH. Effects of moisture content and compression pressure of various deforming granules on the physical properties of tablets. Powder Technol. 2017;310(1):92-102. [CrossRef] | [Google Scholar]
- . Tech Regul Considerations Pharm Prod Lifecycle Manag. 2019;Q12 [CrossRef] | [Google Scholar]
