ABSTRACT
Clinical Trial Monitoring is the fundamental necessity to direct high-quality clinical research to guarantee research quality and subject protection according to regulatory standards. It is a sophisticated, innovation-based approach to utilizing a company’s data, allowing for better-informed decisions regarding where and how to allocate resources. However, the monitoring plan is grouped into two kinds i.e., on-site and centralized monitoring. But recently, regulators have been urged to take on a risk-based monitoring system, as such sponsors are preferring centralized monitoring from the beginning of the trial. Risk-based monitoring assures the quality of trials by managing risks. Furthermore, the Food and Drug Administration (FDA) and European Medicine Agency (EMA) have embraced the risk-based approach and released a guideline paper on the subject. The FDA’s proposed advice from 2013 discusses how to oversee clinical trials using a risk-based approach. Still, on-site monitoring, which is carried out by site visits, and centralized monitoring, which is carried out remotely or offsite, have their self-worth and advantages. Because of the greater complexity of grasping those monitoring procedures, in this review article, we will discuss both monitoring plans with an emphasis on centralized monitoring, which will aid in a better understanding of the entire in a nutshell. Also, we will emphasize the benefits, future, and implementation of Artificial Intelligence/Machine Learning in the risk-based monitoring strategy.
INTRODUCTION
Clinical trial monitoring is the process of supervising the activities of a clinical study for human use medicines and ensuring that they are carried out, in compliance with the protocol, standard operating procedures, and Good Clinical Practices (GCP) Standards, data was collected and reported. The primary goal of monitoring is to ensure that human rights and well-being are protected. It also ensures compliance with regulatory requirements by reviewing the accuracy and completeness of documentation or protocols.1 Effective clinical trial monitoring is pivotal to performing high-quality research while safeguarding subjects involved in this particular research. It comprises different techniques to maintain the same. According to the FDA, a trial monitoring plan is differentiated into two types i.e., onsite monitoring and centralized monitoring, as illustrated in Figure 1.2 However, as per Regulatory Agency encouragement, sponsors have recently adopted a risk-based approach to the monitoring process. As a result, sponsors prefer the centralized monitoring plan from the beginning of clinical trials, which incorporates a risk management strategy.3 The study’s main objective is to demonstrate the distinct features of both onsite and centralized monitoring methods while emphasizing centralized monitoring as an aspect of risk-based monitoring (RBM), which will aid in a better understanding of the overall picture as well as the risk-based approach in monitoring. In the first two sections, we will discuss centralized and on-site monitoring as well as a comparison of both. In the third section, we will discuss the advantages of the use of Artificial Intelligence/ Machine Learning in monitoring. At last, we will provide the future perspective of RBM concisely (Figure 1).
RBM ensures the quality of clinical trials by identifying, monitoring, assessing, and mitigating risks that may have an impact on the study’s quality or safety. The RBM initiative was launched in 2012 as one of five initial projects of Tran’s celebrate aimed at developing efficient and effective solutions. It is a sophisticated, technology-based method for making the best use of a company’s data, allowing it to make more informed decisions about where and how to allocate resources. However, as with many topics, the types of clinical trial monitoring procedures can become muddled or even more complex, and this misunderstanding is often associated with the distinction between on-site versus centralized monitoring.4 On-site monitoring entails in-person review performed at the investigation site by sponsor employees or representatives, whereas Centralized monitoring entails analytical evaluation performed by sponsor employees or representatives at a central location other than the site of the clinical research.5 The primary European and American regulatory agencies like FDA, EMA, and Medicines and Healthcare products Regulatory Agency (MHRA), have praised centralized monitoring as the most appropriate and cost-effective strategy for achieving a higher degree of data quality and enhanced oversight of patient safety. In 2013, the FDA has already released draught guidance on clinical trial oversight, stressing centralized monitoring as part of a risk-based monitoring approach.2 Also, EMA has released a reflection paper on Risk-based quality management in clinical trials.6 On-site monitoring of ongoing clinical trials is a relatively new practice in India. Traditionally, on-site monitoring has been used in the pharmaceutical industry to analyze site performance and mitigate site risk. This method is carried out by someone who frequently visits the trial site. Since the release of their GCP guidance documents in 2011-2013, the agencies have urged clinical researchers to monitor on a risk-based basis.7
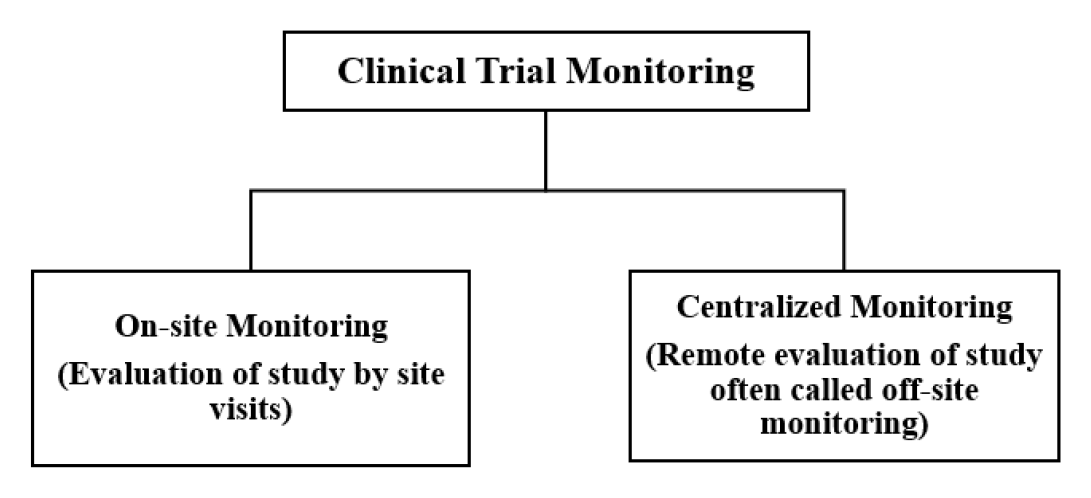
Figure 1.
Types of Clinical Trial Monitoring.
Clinical trial monitoring
Onsite monitoring
On-site monitoring is used in trials all around the world, although techniques vary greatly and there is little evidence to back the practice. These on-site monitoring procedures, including expenditures, must be empirically analyzed to offer strong evidence for the contribution of site visits to trial performance and quality. The monitor can even go so far as to compare the patients’ signatures to those on other documents to ensure they signed the agreement with their consent. The monitor can also compare operational reports to the dates and/or timings of other test results to ensure that no research activities were undertaken without prior authorization. Monitors can examine how the informed consent method with researchers works, as well as any additional consenting source documentation, to ensure that the actions were carried out in line with federal, state, and local laws. Furthermore, the monitor can completely check all medical data, whether paper or electronic, to confirm that each patient fits the criteria for inclusion or exclusion. While analyzing regulatory documentation, the monitor can certify that the site complies with Federal Regulations and the site’s Institutional Review Board (IRB), as well as that the site has obtained all of the necessary documents to conduct the study. With onsite monitoring, a full inventory of documents can be completed, misplaced or lost documentation can be corrected, and regulatory documentation may be reorganized. The monitor can also analyze how well the research staff at the location understands their Institutional Review Board policies and study agreements.8
During product accountability in on-site monitoring, the monitor can determine whether the product is stored in a secure, closed location as required by the sponsors. He also ensures that the sponsor’s merchandise was delivered to the site and appropriately recorded. In addition, the monitor can conduct regular inventory checks to ensure that no product is misplaced or used on non-study Protocol deviations may or may not have an impact on patient safety; The on-site monitor, on the other hand, can cooperate with the site to report major deviations that endanger the safety and welfare of study participants to the sponsor and, if necessary, the site’s IRB. Repeated protocol infractions may also cause the monitor to retrain on faults or examine onerous protocol requirements. The sponsor in preparation for changes can be an extremely useful tool for any research project. Having on-site monitoring visits, rather than an untraceable identity on an E-mail address, assists the study monitor and the site in establishing a working relationship.9 Throughout the experiment, the monitor is often the first point of contact for the research coordinator or Principal Investigator with any questions. If site personnel meet the monitor in person and consider them “useful” rather than “annoying”, they may be more ready to engage with the monitor and sponsor. As a result of this relationship, the monitor can obtain critical information from the study team that would otherwise go unnoticed. During a casual conversation, the monitor can learn about the site staff’s knowledge of the protocol, regulations, and responsibilities outside of the clinical trial that may prevent them from fully focusing on the trial, as well as their process for obtaining informed consent and assessing adverse events (Figures 2 and 3).10
Benefits of onsite monitoring
Onsite monitoring provides us with the benefit of building a relationship with the study team at the site and checking the basis of data and knowledge which includes: Source data verification (SDV), Correct handling of the investigational medicinal product (IMP), Patient information/Informed consent, Adverse event (AE) reporting, Investigator site file (ISF).
It also benefits us as we can react through involvement: Brosteanu et al. conducted a clustered randomized study comparing intensive onsite monitoring and risk-adapted monitoring in response to growing concern about the effectiveness and efficiency of monitoring procedures. The benefit of intensive onsite SDV was found to be small (8.2%) when compared to risk-based monitoring, which used less than half the resources while ensuring the same level of GCP compliance.10
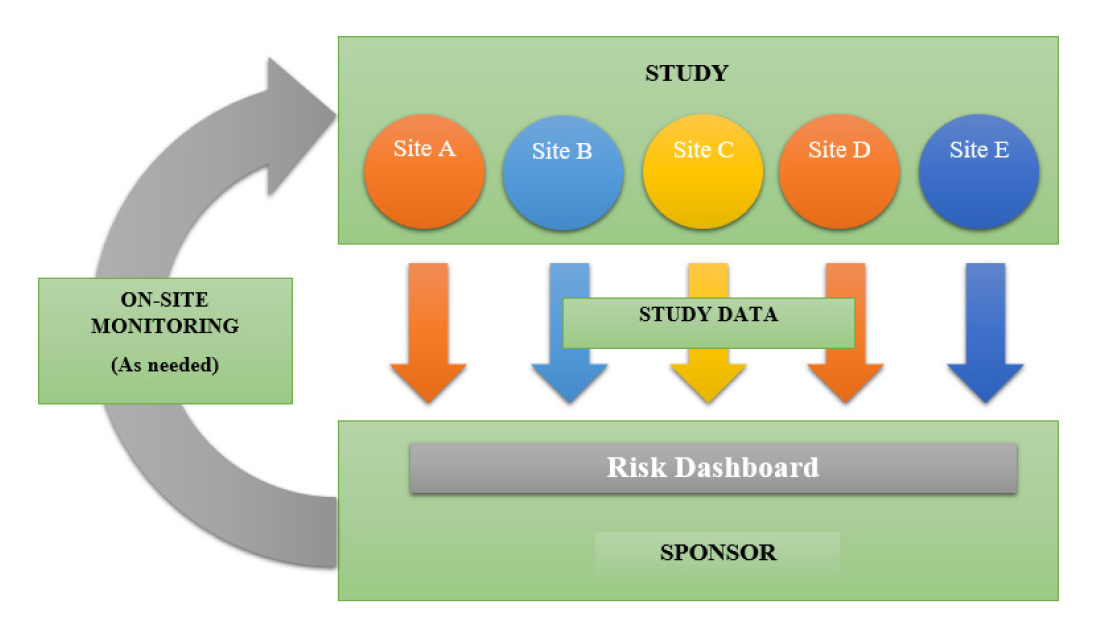
Figure 2.
Onsite monitoring Process.
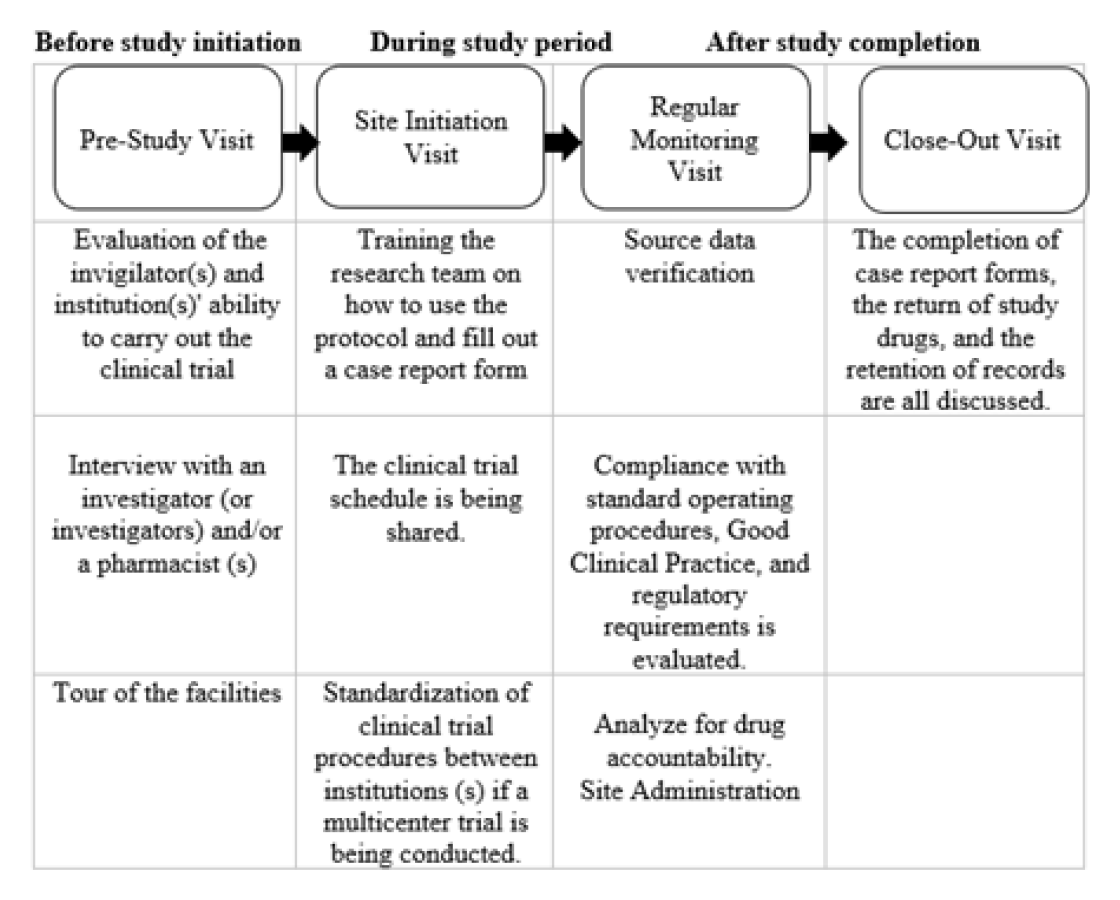
Figure 3.
Onsite monitoring in different stages of a trial.
Centralized monitoring
A remote assessment by persons or representatives from the sponsor (e.g., clinical monitors, data management workers or statisticians) at a site other than the clinical trial location is known as centralized monitoring. Many on-site monitoring capabilities, as well as additional capabilities, can be handled using centralized monitoring procedures.11
The FDA recommends, where appropriate, the use of centralized monitoring procedures, with a corresponding decrease in emphasis on on-site monitoring. Several factors, including the sponsor’s use of electronic systems, access to subjects’ electronic records, if applicable, timeliness of data entry from paper CRFs, if applicable, and communication tools available to the sponsor and study site, determine the types of monitoring activities and the extent to which centralized monitoring practices can be used. These will vary depending on the study and location. Sponsors considering centralized monitoring systems should ensure that processes and expectations for site record-keeping, data entry, and reporting are well specified, and that documentation is easily available.2 If sponsors intend to rely primarily on centralized monitoring procedures, the monitoring plan should specify when one or more on-site monitoring visits are required. “Centralized monitoring is a remote evaluation performed by sponsor people or representatives at a location other than the clinical trial site(s),” according to the most recent draught guidance (Figure 4).
Centralized monitoring techniques should be used to the extent appropriate and feasible to ‘Complement or reduce the frequency and scope of on-site monitoring with monitoring activities that can be performed just as well or better remotely, or with monitoring activities that can only be performed through centralized processes. Review submitted data regularly to identify and follow up on missing data, inconsistent data, data outliers, and potential protocol deviations that could indicate systemic or substantial errors in data collection and reporting at a site of record, for example.12
Conduct statistical studies to uncover data trends that are difficult to detect through onsite monitoring, such as standard checks of the data range, consistency, and completeness, as well as checks for unusual data distribution within and between study sites, such as little variances. In instances where such source data is accessible or if CRF data is, according to the protocol, source data, complete administrative and regulatory duties by remotely checking important source data as defined in the monitoring plan.13
‘Constantly verifying (IRB) approval, such as by examining electronic IRB correspondence if available; determining whether previously requested CRF adjustments were made; and performing investigational product accountability tasks, such as comparing randomization and CRF data to determine whether the subject was administered or dispensed the assigned product, and assessing consistency between investigational product receipt, use, and disposal. Routine reviews of provided data, as well as statistical and other analytics, can be utilized to spot major problems. (e.g., the need for clarification of a protocol procedure, indications of data fabrication) with non-critical data that would not otherwise have been a focus of monitoring (e.g., source document verification).7
Benefits of centralized monitoring
Centralized Monitoring provides us with the benefit as it assists in the early detection and mitigation of data quality risk/issue(s) that may jeopardize the validity of study findings; Contributes to improved patient safety monitoring and subject protection; Improves CRA performance by allowing them to use their time more efficiently and objectively while on-site monitoring; Monitors site performance to assist in the planning of on-site monitoring visits. Improves on-site monitoring visits while lowering associated costs. As a result, overall clinical trial activities are more efficient.14 The comparison between Onsite and Centralized monitoring is provided in Table 1.
Benefits of RBM, Integration of AI/ML in Monitoring Plan
In this section, the benefits of RBM are clearly defined. Also, the integration of AI/ML for the improvement of the monitoring plan is provided in a brief.
Benefits of Risk-based monitoring
Risk-based monitoring provides us with the benefit of cost savings as monitoring efforts account for more than a quarter of a clinical trial’s total cost. As a result, by optimizing monitoring, we can save a lot of money; Faster, more focused results as data analysis and centralized monitoring allow for more accurate and timely findings; RBM research reduced the number of missing pages by 45 percent, and the critical data error rate by four times when compared to traditional studies; Real-time data entry, RBM encourages real-time data entering. Sites that employ RBM are 5 times more likely to enter trial data within 7 days, according to comparison studies; Enhanced emphasis on the primary goal as the administrative activities are greatly reduced, giving employees more time to focus on their core roles and responsibilities; Increased participant safety, by reducing administrative activities, resources can be better allocated and focused on assuring participant safety; Better compliance, Data-driven insights, and other digital tools help with early discovery of problems, site training, and the detection of potentially fraudulent actions. These variables inevitably lead to increased compliance; Increased collaboration as RBM encourages coworkers to collaborate and share more.15
| Reviewed Items | Centralized Approach | On-site Approach |
|---|---|---|
| Informed consent | Examine the documents of the site’s signed informed consent agreements. Verify that the consent contains all required components, which the correct version of the consent is used, and that signatures are included. | Examine source documents to verify the patient’s signature, that the patient gave his or her consent prior to research operations, and that the site followed the requisite consent procedures. |
| Eligibility criteria | Verified according to the source supplied by the site. | All available medical records (paper and/or electronic) were subjected to a source-verified review. |
| Protocol procedures | Examine the Case Report forms (CRFs)/sources sent by the site to check compliance with the protocol. | CRF source verification, capacity to support the site with question resolution, obtaining outstanding data, and so forth. Determine the site’s level of comfort with procedure requirements through discussion. |
| Regulatory documents | Manage a “shadow file” of the site’s regulatory documents, according to information received from the sites. | Examine all regulatory documentation to ensure that records are accurate, up to date, and complete. Assist with the correction of any errors or missing documents on the site. Verify that the sponsor has a copy of the necessary paperwork. |
| Investigator involvement | Discussions by phone or e-mail to check on the status of the study or to answer questions. | In-person interactions enable the monitor to identify critical site/staff issues that would not be apparent otherwise. |
| Product accountability | Checking the product log against the facility’s shipping records and, possibly, product labels on patient source documents. | Verifying product log against sponsor shipping records, patient source device labels, products in stock, and product storage location. |
| Adverse events | Remote evaluation of AEs submitted by the site. | Review of all accessible medical records (paper and/or electronic) for additional unreported adverse events; conversation with site concerning adverse event reporting to the IRB. |
| Protocol deviations | Remote protocol variations supplied by the site are reviewed. | Examine the site for any unreported protocol violations, talk to the site about reporting deviations to the IRB, and see if extra training is needed. |
| Relationship with site staff | Communication is limited when only telephone and e-mail are used. | In-person visits allow the monitor/sponsor to put a face to the name, which may lead to a more comfortable working relationship. |
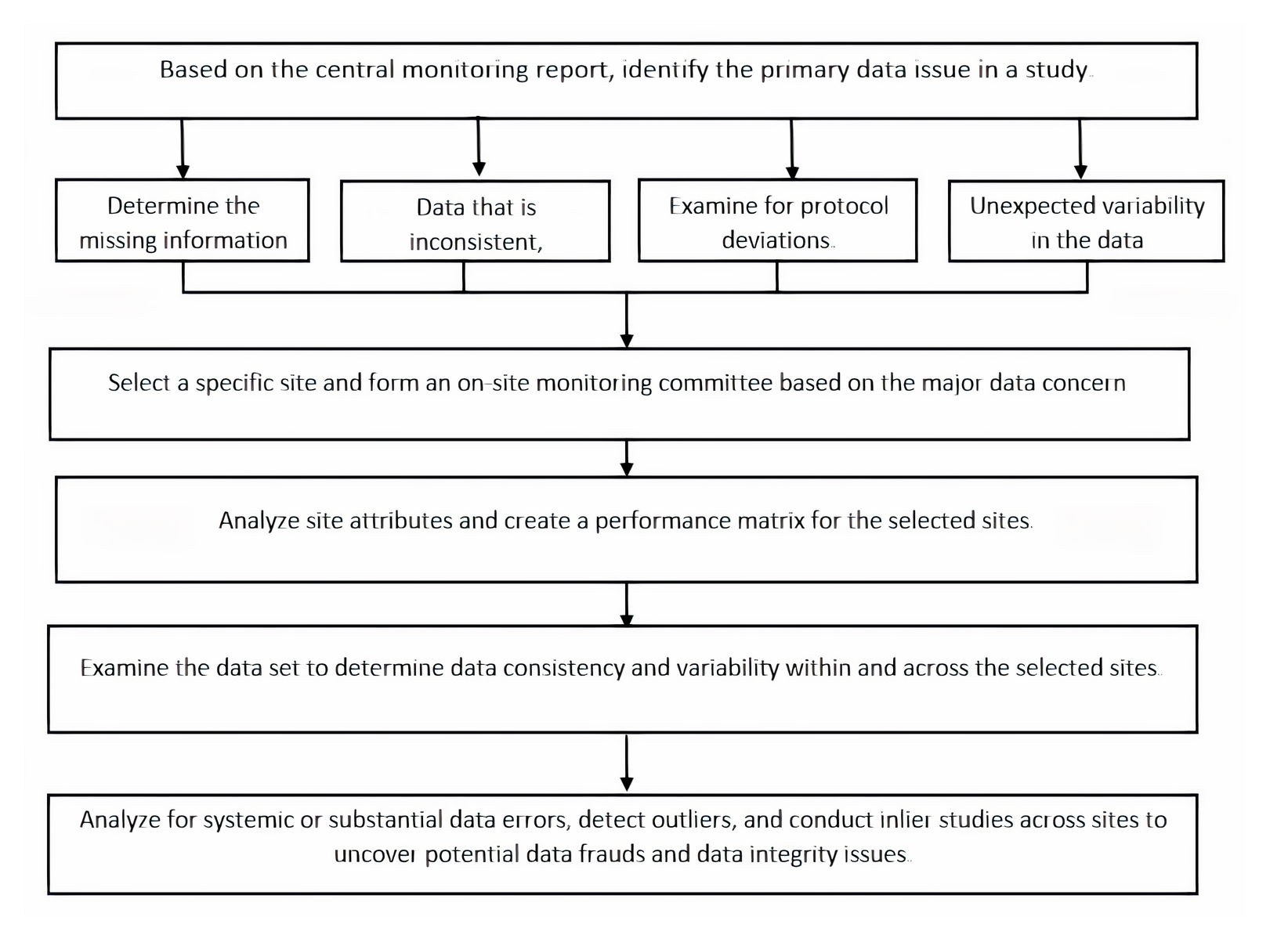
Figure 4.
Flow chart for the centralized data review process.
Use of AI/ML in the risk-based monitoring process
AI/ML is currently being employed extensively in the pharmaceutical and healthcare industries, and it is also having a significant impact on clinical trials because of the adaptation of electronic health records, consumer safety reports, etc.16 Deep learning (DL) techniques, which are part of AI/ML, can be used to fine-tune the model based on new data obtained from ongoing studies, and predictive analytics, which is used to determine trial risk, can be used in centralized/risk-based monitoring to assess investigator site performance and predict it based on previous performance. This study could be utilized to develop adaptive site monitoring strategies for high-risk locations, such as increasing monitoring frequency and resource allocation. Automatic learning of temporal dependence and automatic handling of temporal structures like trends and seasonality, which may be used to train the model and offer more accurate forecasts, are also possible with DL techniques for Time Series forecasting. Risk assessment and resource allocation.17
Future Perspective
Nobody knows what will happen in the future. Predictions appear to be speculative at best. One thing is certain: the way clinical trials are carried out will change. In recent years, there has been a significant shift toward patient-centricity, adaptive designs, synthetic control arms, and decentralized trials. These developments are likely to continue, as there is a push to shorten development cycle times. Modern RBM technologies and methodologies have the potential to enhance the drug development landscape.18 Adopting appropriate RBM procedures and technology can help sponsors improve research predictability, patient safety, and data quality by determining where and how to change their operations to address future issues. It is also one method of combating the ever-increasing costs of conducting good clinical research. Limited resources and funds can be used more effectively to ensure data quality by shifting costs away from on-site inspections and 100 percent Source Data Verification (SDV) and toward centralized data collection and monitoring. While the challenges of switching to a different monitoring technique have hampered industry-wide adoption, RBM’s future appears bright. Progress is being made, and key stakeholder engagement is becoming more common.19 At most, the traditional site model and centralized monitoring approaches will have to change as the drug development scenario changes significantly, including the type of investigational medicines, increased use of technology such as eSource such as e-health records, wearables such as smart monitoring devices, and the integration of AI/ML.17
CONCLUSION
As is always the case, no single monitoring strategy is optimal for any clinical research. The monitoring method must be adapted and documented in the monitoring plan based on the study’s features (including risks). Onsite monitoring as the traditional practice is cost-consuming and somewhat complicated as the monitor has a great responsibility, for example, managing, reviewing, and evaluating the documents conventionally. Still, it has a lot of advantages like relationship building, reviewing subject consent, and determining the misplaced documents. Also, this methodology is extremely valuable during investigational medicinal product research. On the other hand, centralized monitoring, a part of risk-based monitoring, plays a crucial role in limiting costs and resources by detecting risks right off the bat. However, in most circumstances, a (risk-based) mix of centralized/remote and on-site monitoring is a suitable solution because both types have their value. The risk-based strategy is popular because of modernization, and regulatory adaptation by different. It is beneficial to identify and reduce risks at an early stage, which facilitates the shift in drug development. Lowering the costs, faster results, real-time data entry, complying with safety with lower error, and better compliance are some of the benefits of RBM which attract the sponsors to integrate it into clinical trials. The application of AI/ML has the potential to improve the performance of risk-based monitoring and will aid in greater compliance shortly. As RBM is the most efficient approach, it has a strong future.
Cite this article
Verma V, Mishra A, Gowrav MP, Nischitha MS. Clinical Trial Monitoring: An Overview of Risk-based Approach. J Young Pharm. 2023;15(2):239-44.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
References
- [cited Oct 30 2022];Handbook for good clinical research practice (gcp). Who.int. Available from:https://apps.who.int/iris/bitstream/handle/10665/43392/924159392X_eng.pdf?sequence=1&isAllowed=y
[Google Scholar] - [cited Jul 1 2022];Guidance for industry oversight of clinical investigations – A risk-based approach to monitoring. Fda.gov. 2013 Available from:https://www.fda.gov /media/116754/download
[Google Scholar] - SOCRA. [cited Jul 1 2022];The value of centralized monitoring [internet]. SoCRA blog. 2019 Available from:https://www.socra.org/blog/the-value-of-centralized-monitoring/
[Google Scholar] - Celerate. [cited Jul 1 2022];Risk based monitoring [internet]. 2015 Translator Available from:https://www.transceleratebiopharmainc.com/initiatives/risk-based-monitoring/
[Google Scholar] - Butler R. [cited Jul 1 2022];Understanding the 3 types of clinical trial monitoring [internet]. Pharm-olam.com. Available from:https://www.pharm-olam.com/blog/3-types-of-clinical-trial-monitoring
[Google Scholar] - [cited Jul 1 2022];Reflection paper on risk-based quality management in clinical trials Table of contents. Europa.eu. 2013 Available from:https://www.em a.europa.eu/en/documents/scientific-guideline/reflection-paper-risk-based-quality-management-clinical-trials_en.pdf
[Google Scholar] - Andrianov A. [cited Jul 1 2022];Centralized Monitoring: A greater advantage to a broader range of trials [internet]. Cyntegrity.com.Cyntegrity. 2019 Available from:https://cyntegrity.com/centralized-monitoring-in-clinical-research/
[Google Scholar] - Agrafiotis DK, Lobanov VS, Farnum MA, Yang E, Ciervo J, Walega M, et al. Risk-based monitoring of clinical trials: An integrative approach. Clin Ther. 2018;40(7):1204-12. [PubMed] | [CrossRef] | [Google Scholar]
- [cited Jul 01 2022];Imarcresearch.com. Available from:https://www.avaniaclinical.com/?s=Centralized+vs+Onsite+Monitoring%3A+A+Sponsor%E2%80%99s++Bala ncing+Act+Applying+a+Risk-based+Approach
[PubMed] | [CrossRef] | [Google Scholar] - Jung SY, Kang JW, Kim TH. Monitoring in clinical trials of complementary and alternative medicine. Integr Med Res. 2021;10(2):100666 [PubMed] | [CrossRef] | [Google Scholar]
- Houston L, Probst Y, Yu P, Martin A. Exploring Data Quality Management within Clinical Trials. Appl Clin Inform. 2018;9(1):72-81. [PubMed] | [CrossRef] | [Google Scholar]
- Risk-based Monitoring. [cited Jul 1 2022];The next revolution in Clinical trials [internet]. Acceliant.com. Available from:https://www.acceliant.com/articles/risk-based-monitoring-the-next-revolution-in-clinical-trials
[PubMed] | [CrossRef] | [Google Scholar] - Afroz MA, Schwarber G, Bhuiyan MAN. Risk-based centralized data monitoring of clinical trials at the time of COVID-19 pandemic. Contemp Clin Trials. 2021;104(106368) [PubMed] | [CrossRef] | [Google Scholar]
- Beauregard A, Labrie F, Tantsyura V. The basics of clinical trial centralized monitoring [serial on the Internet]. Appl Clin Trials. 2018 [PubMed] | [CrossRef] | [Google Scholar]
- Saama technologies. [cited Jul 1 2022];Risk Based Monitoring: What is it and How Can You Benefit [serial on the Internet]. Inc. Saama.com. Saama technologies. 2017 Available from:https://www.saama.com/risk-based-monitoring-what-is-it-and-how-can-you-benefit/
[PubMed] | [CrossRef] | [Google Scholar] - Mishra A, Gowrav MP, Balamuralidhara V, Reddy KS. [cited Jul 1 2022];Health in digital world: A regulatory overview in United States. J Pharm Res Int [serial on the Internet]. 2021:438-50. 2021 Available from:https://journaljpri.com/index. php/JPRI/article/view/32573
[PubMed] | [CrossRef] | [Google Scholar] - Vyas NR. Future of risk based monitoring in clinical trials. Int J Clin Trials. 2020;7(3):221 [CrossRef] | [Google Scholar]
- Barnes B, Stansbury N, Brown D, Garson L, Gerard G, Piccoli N, et al. Risk-based monitoring in clinical trials: Past, present, and future. Ther Innov Regul Sci. 2021;55(4):899-906. [PubMed] | [CrossRef] | [Google Scholar]
- [cited Jul 1 2022];Risk-based monitoring: Understanding the future of clinical trial monitoring [internet]. Clinicalleader.com. Available from:https://www.clinicalleader.com/doc/risk-based-monitoring-understanding-the-future-of-clinical-trial-monitoring-0001
[PubMed] | [CrossRef] | [Google Scholar] - [cited Jun 28 2022];SOP: 3. Clinical trial monitoring [internet]. Khpcto.co.uk. Available from:https://khpcto.co.uk/SOPs/03_MonitoringSOP.php
[PubMed] | [CrossRef] | [Google Scholar]
